SoybeanGDB is a comprehensive database providing informatics service to researchers focused on the genetic and genomic studies of soybean. A total of 39 high-quality soybean genomes were collected, including a Chinese soybean Glycine max [L.] Merr. cv. Zhonghuang 13. High-quality SNPs and INDELs among 2898 soybean accessions based on the genome of Zhonghuang 13. High-quality SNPs among 481 soybean accessions based on the genome of Williams 82.
 SoybeanGDB:
a comprehensive genome database of soybean
SoybeanGDB:
a comprehensive genome database of soybean
SoybeanGDB:
a comprehensive genome database of soybean
JBrowse of 39 soybean genomes
Calculation In progress...

Calculation In progress...
Search one or multiple soybean genomes by sequence similarity using BLAST
Information of selected soybean accessions
Information of selected soybean accessions
39 soybean genomes
SNPs among 2898 soybean accessions
https://ngdc.cncb.ac.cn/gvm/getProjectDetail?project=GVM000063
SNPs among 481 soybean accessions
https://doi.org/10.1038/s41597-021-00834-w
Expression profile of protein-coding genes in the genome of Zhonghuang 13
https://link.springer.com/article/10.1007/s11427-019-9822-2
Expression profile of protein-coding genes in the genome of A81-356022
https://bmcplantbiol.biomedcentral.com/articles/10.1186/1471-2229-10-160
Expression profile of protein-coding genes in the genome of W05
https://www.nature.com/articles/ncomms5340
Transcriptomes data of 102 soybean accessions
https://bmcplantbiol.biomedcentral.com/articles/10.1186/1471-2229-10-160
SoybeanGDB
- Source code: github.com/venyao/SoybeanGDB
- Online use: venyao.xyz/SoybeanGDB/
- Contact: yaowen@henau.edu.cn
Related tools and databases
- BLAST: Basic Local Alignment Search Tool
- Tabix: Generic indexer for TAB-delimited genome position files
- Primer3: A widely used program for designing PCR primers
- CNCB-NGDC: China National Genomics Data Center
- SoyBase: The USDA-ARS soybean genetics and genomics database
- RepeatMasker: A program that screens DNA sequences for interspersed repeats and low complexity DNA sequences
- OrthoFinder: A program for phylogenetic orthology inference
- JBrowse2: A pluggable open-source platform for visualizing and integrating biological data
- Wildsoydb: Genetic resources for legumes
- ECOGEMS: Efficient compression and retrieve of SNP data of 2058 rice accessions with integer sparse matrices
- MaizeSNPDB: A comprehensive database for efficient retrieve and analysis of SNPs among 1210 maize lines
Related publications
- Pan-Genome of Wild and Cultivated Soybeans
- A new decade and new data at SoyBase, the USDA-ARS soybean genetics and genomics database
- A reference-grade wild soybean genome
- The genome portal of the Department of Energy Joint Genome Institute: 2014 updates
- NCBI BLAST: a better web interface
- Primer3—new capabilities and interfaces
- JBrowse: a dynamic web platform for genome visualization and analysis
- clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters
- eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses
- Tabix: fast retrieval of sequence features from generic TAB-delimited files
- SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies
- Soybean DICER-LIKE2 Regulates Seed Coat Color via Production of Primary 22-Nucleotide Small Interfering RNAs from Long Inverted Repeats
- Phylogenomics of the genus Glycine sheds light on polyploid evolution and life-strategy transition
- Genome assembly of the popular Korean soybean cultivar Hwangkeum
- Genetic variation among 481 diverse soybean accessions, inferred from genomic re-sequencing
- RNA-Seq Atlas of Glycine max: A guide to the soybean transcriptome
Tutorial of SoybeanGDB
SoybeanGDB is a comprehensive genome database to accelerate functional genomic and population genetic studies in soybean.
SoybeanGDB employs 39 high-quality soybean genomes, 15,446,616 high-quality SNPs (single nucleotide polymorphism) and 4,136,231 high-quality Indels (small insertions/deletions) among 2898 soybean accessions identified based on next-generation sequencing data. To help users, a variety of versatile analytic tools including JBrowse, BLAST, GO/KEGG annotation, GO/KEGG enrichment analysis, Primer designing, etc., are implemented in SoybeanGDB. SoybeanGDB is deployed at (https://venyao.xyz/SoybeanGDB/) for online use.
The homepage of SoybeanGDB displays the main functionalities of SoybeanGDB (Figure 1).
(1) Search 39 high-quality soybean genomes by gene IDs.
(2) Search 39 high-quality soybean genomes by genome locations.
(3) Search 39 high-quality soybean genomes using BLAST.
(4) Browse transcription factors/regulators in a genome.
(5) Browse syntenic regions between different soybean genomes.
(6) Browse structural variations by location.
(7) JBrowse visualization of 39 high-quality soybean genomes.
(8) Browse and visualize high-quality SNPs among 2898 soybean accessions.
(9) Search and retrieve SNPs among 2898 soybean accessions.
(10) Conduct linkage disequilibrium analysis between SNPs in a genomic region.
(11) Conduct nucleotide diversity analysis among different groups of soybean accessions to identify genes under selection during domestication and modern breeding.
(12) Calculate and visualize allele frequency of user-input SNP sites.
(13) Search and retrieve INDELs among 2898 soybean accessions.
(14) Conduct expression and co-expression analysis of genes in the genome of Zhonghuang 13.
(15) Design primers based on the Zhonghuang 13 genome targeting SNPs and Indels in user-input genomic region.
(16) Search and retrieve orthologous gene groups among 39 soybean genomes.
(17) Conduct GO (gene ontology) annotation/enrichment analysis of user-input protein-coding genes in any of the 39 genomes.
(18) Conduct KEGG annotation/enrichment analysis of user-input protein-coding genes in any of the 39 genomes.
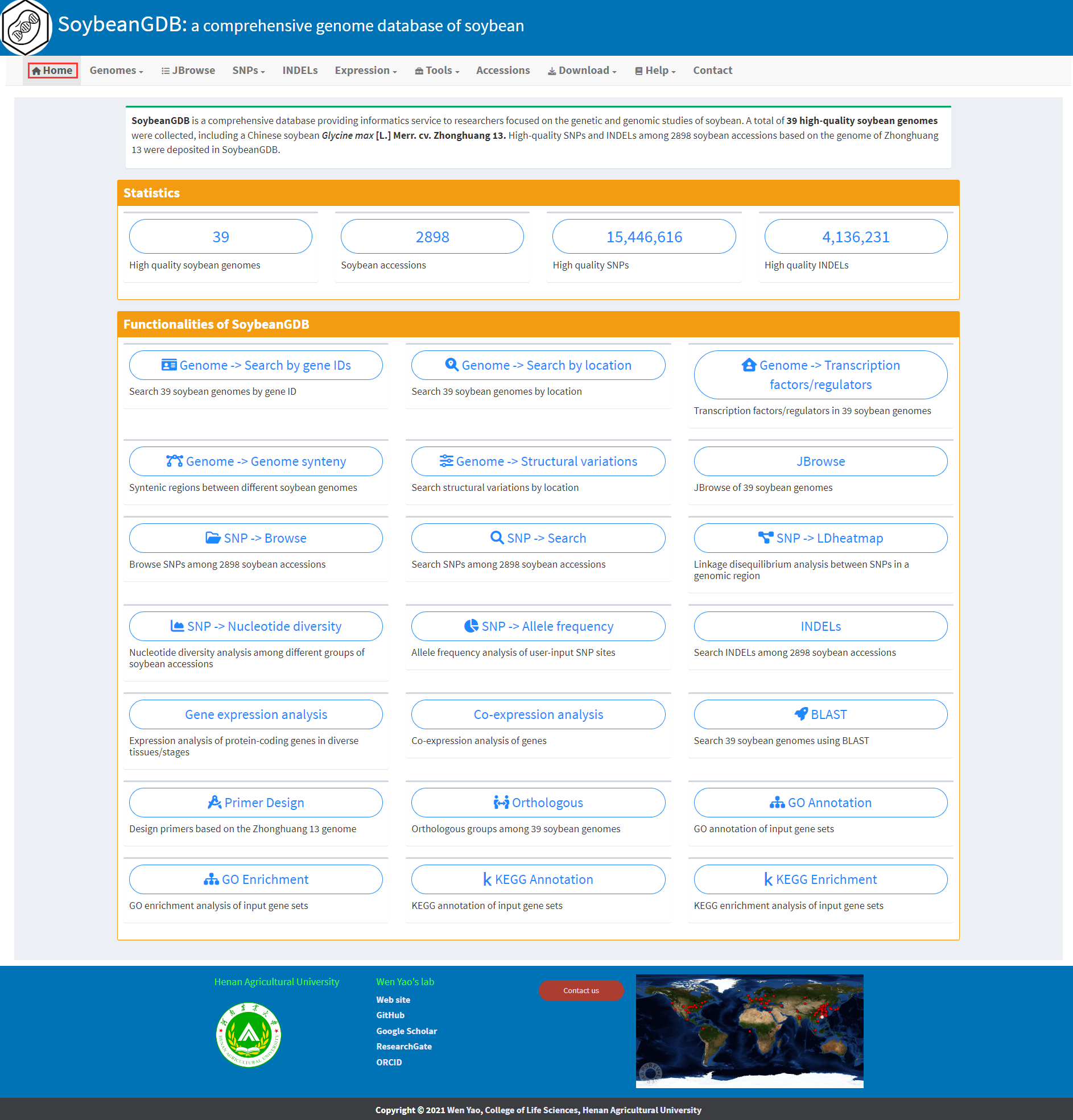
Figure 1. The home page of SoybeanGDB
1. Search a genome by gene IDs
Users can input multiple gene IDs for any of the 39 soybean genomes to obtain the annotations of all input genes. Annotations of all genes can be viewed in a table, which can be exported as a csv or excel file (Figure 2). The gene sequences, CDS sequences, cDNA sequences, and the protein sequences can be download as plain text files.

Figure 2. Search a genome by multiple gene IDs
2. Search a genome by location
Users can input a genomic region to view and retrieve the information of genes and transposable elements in any of the 39 soybean genomes. Check the example input data to enter a genomic region in appropriate format (Figure 3). Steps to search any genome by location are shown in Figure 3.

Figure 3. Genes and transposable elements in chr1:20260371-20686979 of Zhonghuang 13
3. Genomic distribution of genes
Users can input multiple gene IDs for any of the 39 soybean genomes to visualize the chromosome location of all input genes (Figure 4).
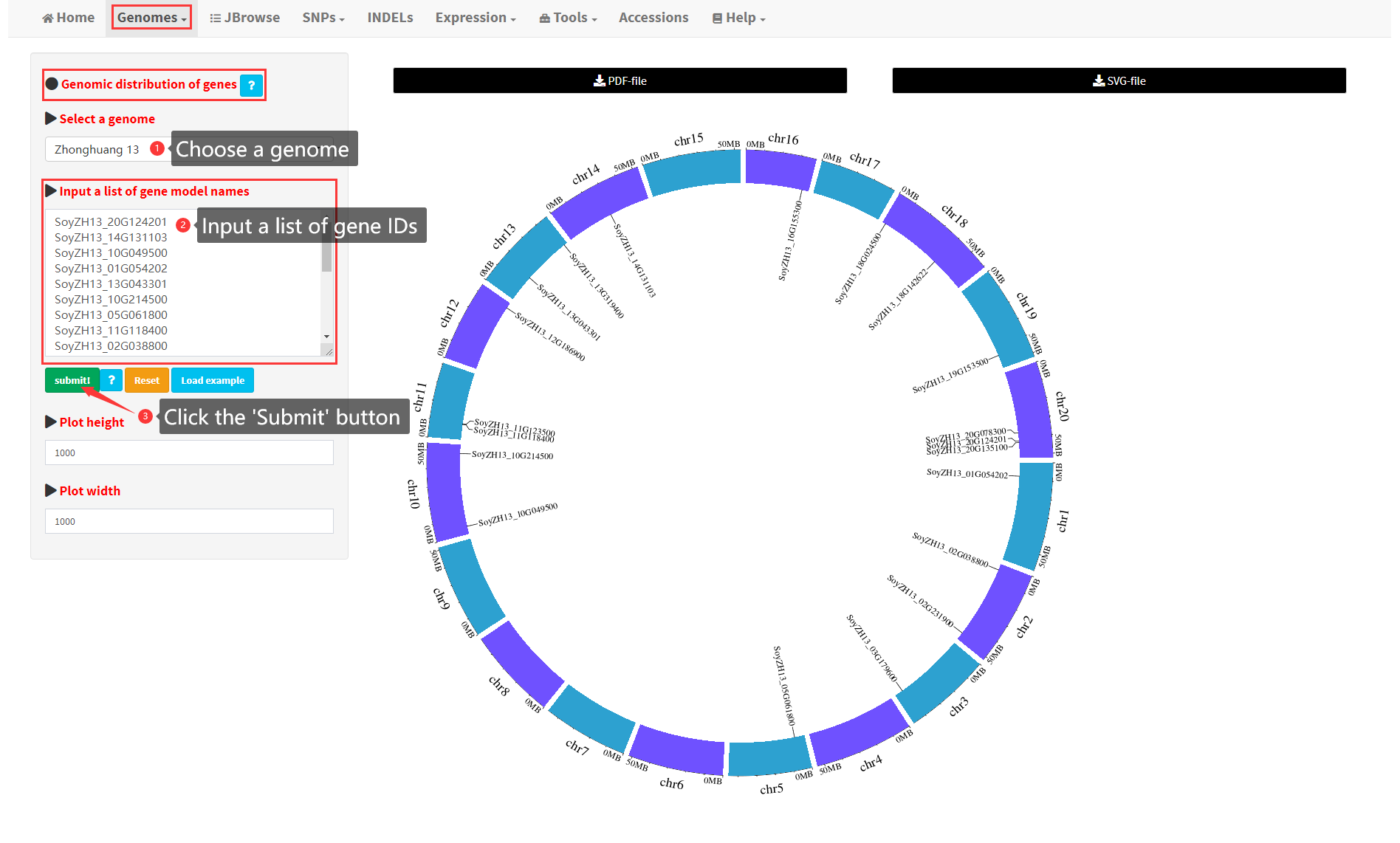
Figure 4. Genomic distribution of genes
4. Browse transcription factors/regulators of a genome
Users can browse transcription factors/regulators annotated in any of the 39 soybean genomes. Steps to browse the annotated transcription factors/regulators are shown in Figure 5.

Figure 5. Transcription factors/regulators of Zhonghuang 13
5. Genome synteny
This function is designed for users to browse the syntenic regions between two soybean genomes. Syntenic blocks will be displayed in a table in the main panel. Genes in the syntenic blocks of the reference/query genome will be listed (Figure 6).
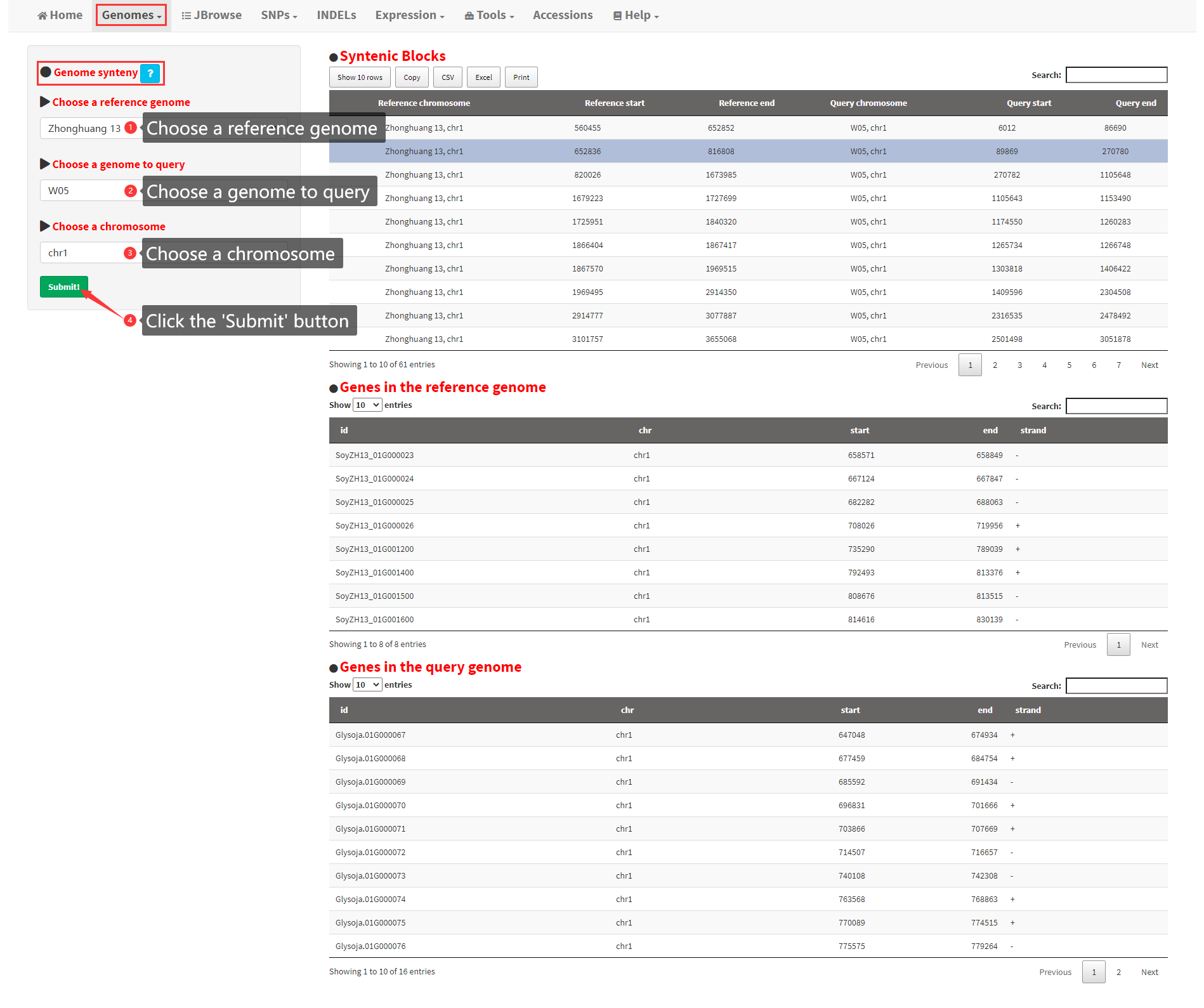
Figure 6. Syntenic chromosome regions between Zhonghuang 13 and W05
6. Structural variations
This function is designed for users to browse the structural variations identified between two soybean genomes. Different types of structural variations, including duplications, inversions, tandem repeats, translocations, deletions, and insertions, will be displayed in different panels (Figure 7).
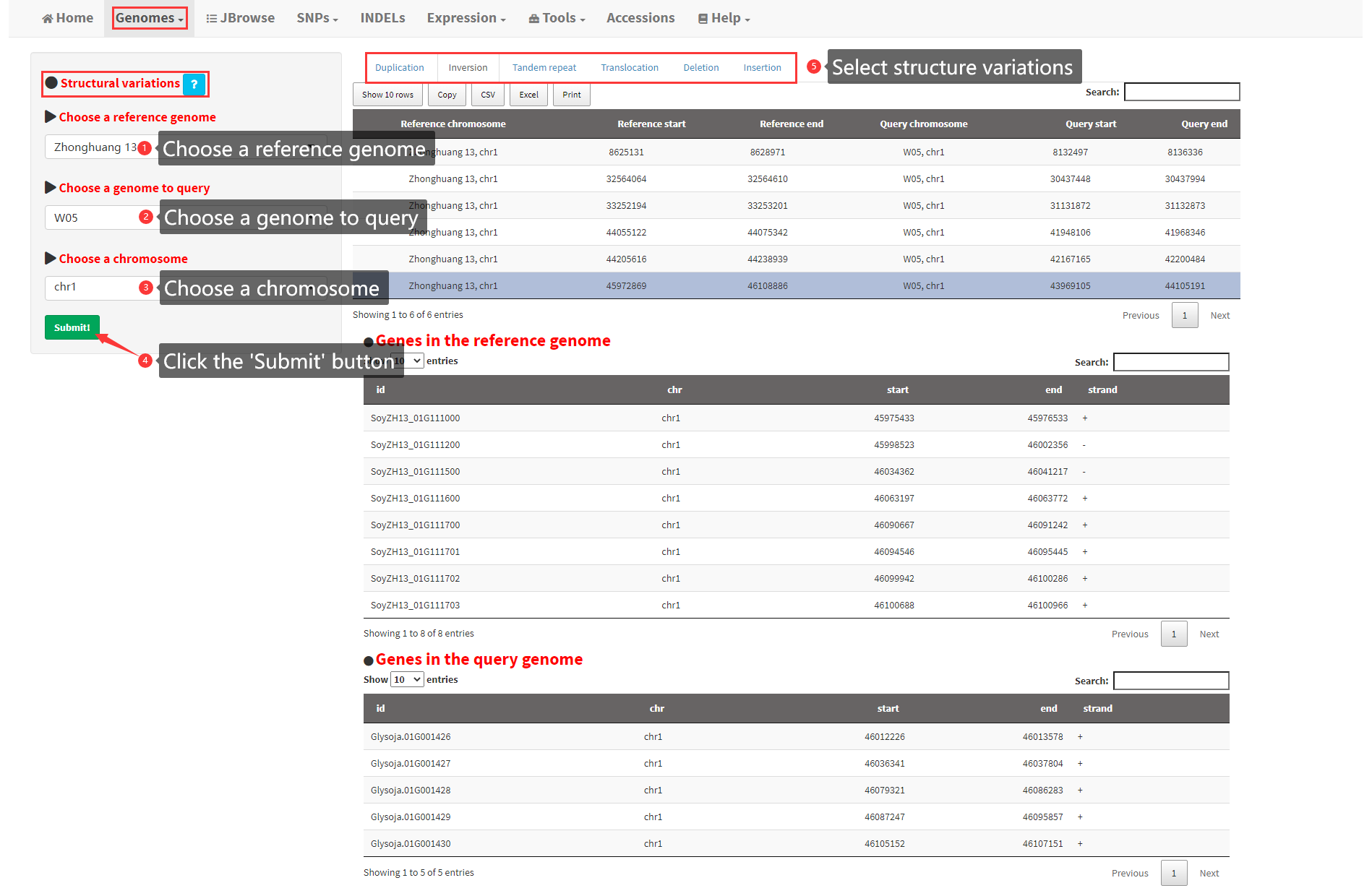
Figure 7. Structural variations between Zhonghuang 13 and W05
7. JBrowse
Jbrowser of all 39 high-quality soybean genomes were built to view the genome sequence, protein-coding genes, transposable elements, as well as the GO and KEGG annotations of protein-coding genes (Figure 8; Figure 9). These information are displayed in different tracks of the JBrowser.
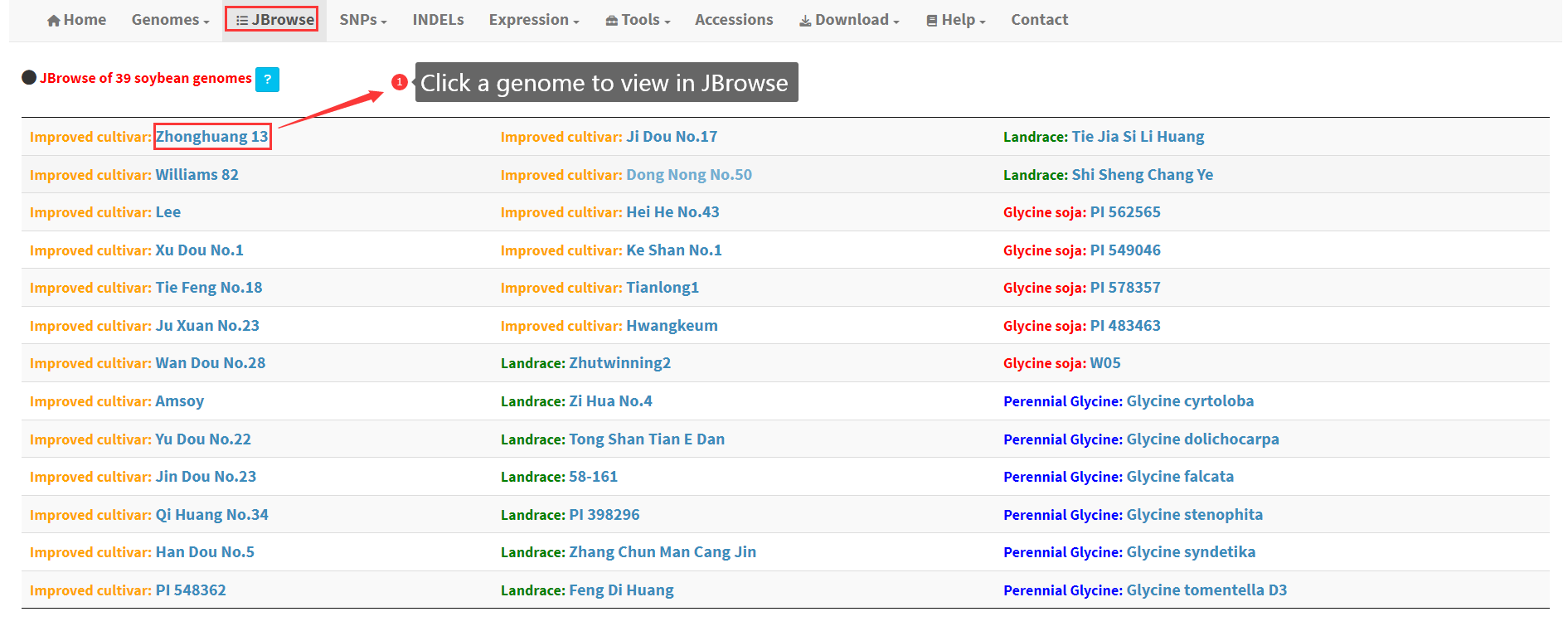
Figure 8. The JBrowse menu of SoybeanGDB
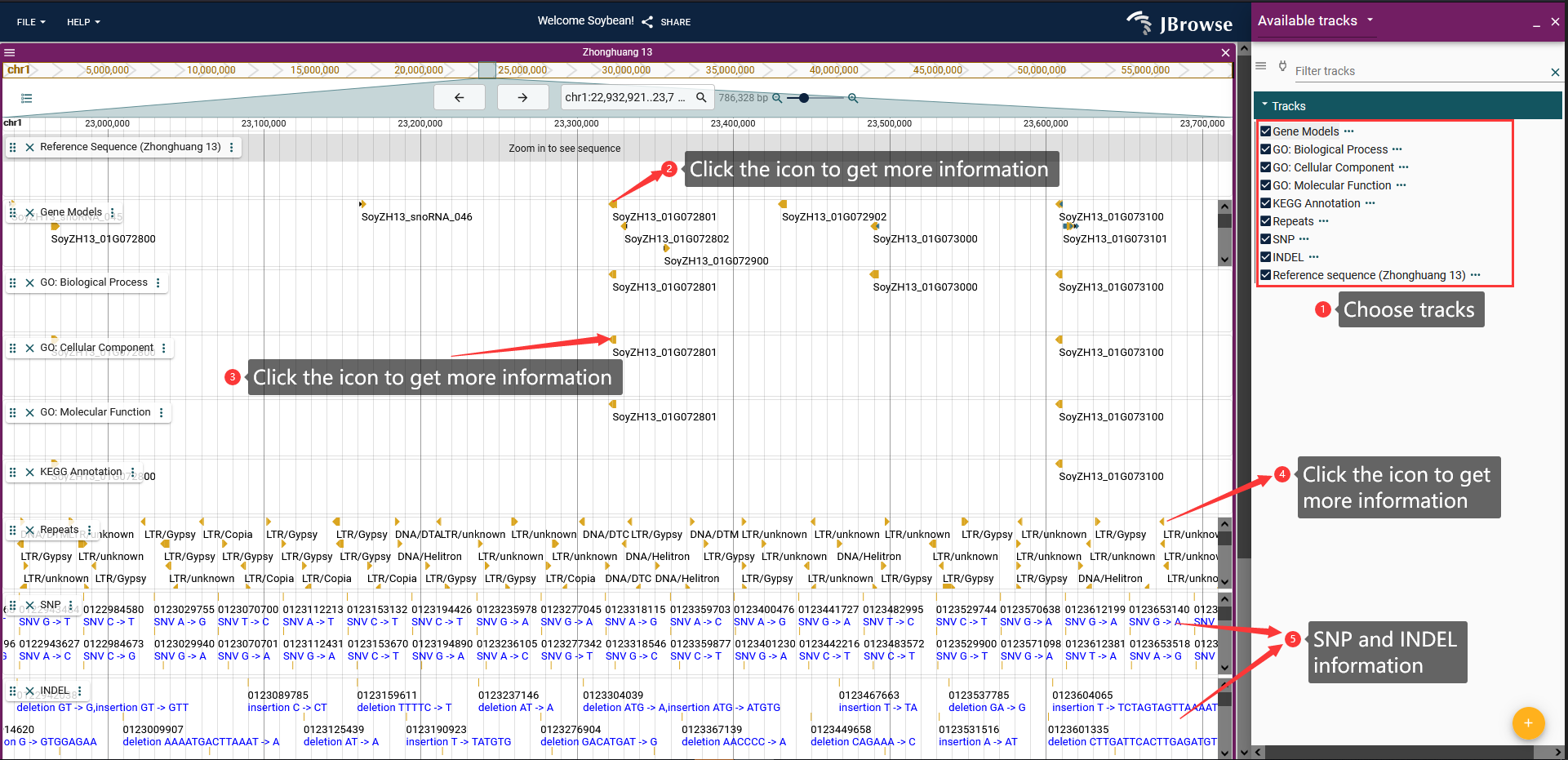
Figure 9. JBrowse of Zhonghuang 13
8. Browse SNPs in a user-input genomic region
Users can browse and download SNPs among 2898 soybean accessions by inputting a single gene ID or genomic region in appropriate format, such as “SoyZH13_09G103313” or “chr1:29765419-29793053” (Figure 10). After clicking the “Submit” button, SNPs will be visualized in the main panel, with different SNPs represented as inverted-triangles in different colors. The result can be further filtered by selecting soybean accessions or setting the mutation effect of SNPs. The result can also be downloaded by clicking the download buttons “Genotype data”, “SNPs information” and “PDF-file” at the top of the main panel.
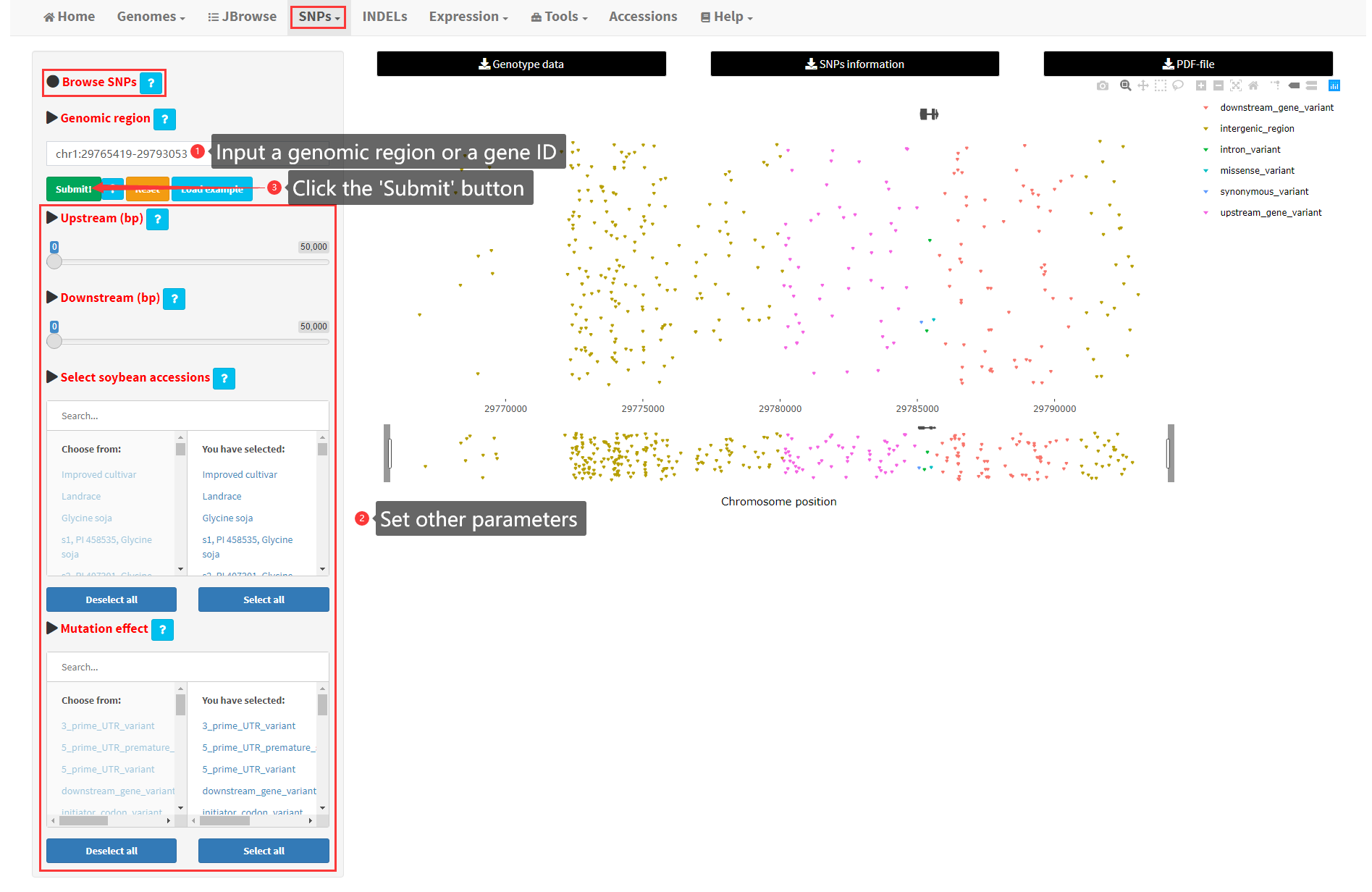
Figure 10. Browse and visualize SNPs in chr1:29765419-29793053
9. Search and retrieve SNPs in a user-input genomic region
Users can search and retrieve SNPs among 2898 soybean accessions by inputting a single gene ID or genomic region in appropriate format, such as “chr7:29560705-29573051” (Figure 11). After clicking the “Submit” button, the genotypes of selected soybean accessions at SNP sites located in the user-input genomic region will be displayed in the main panel. In addition, SNPs information, genotype data and gene annotations in user-input genomic region can be downloaded using the download buttons at the top of the main panel.
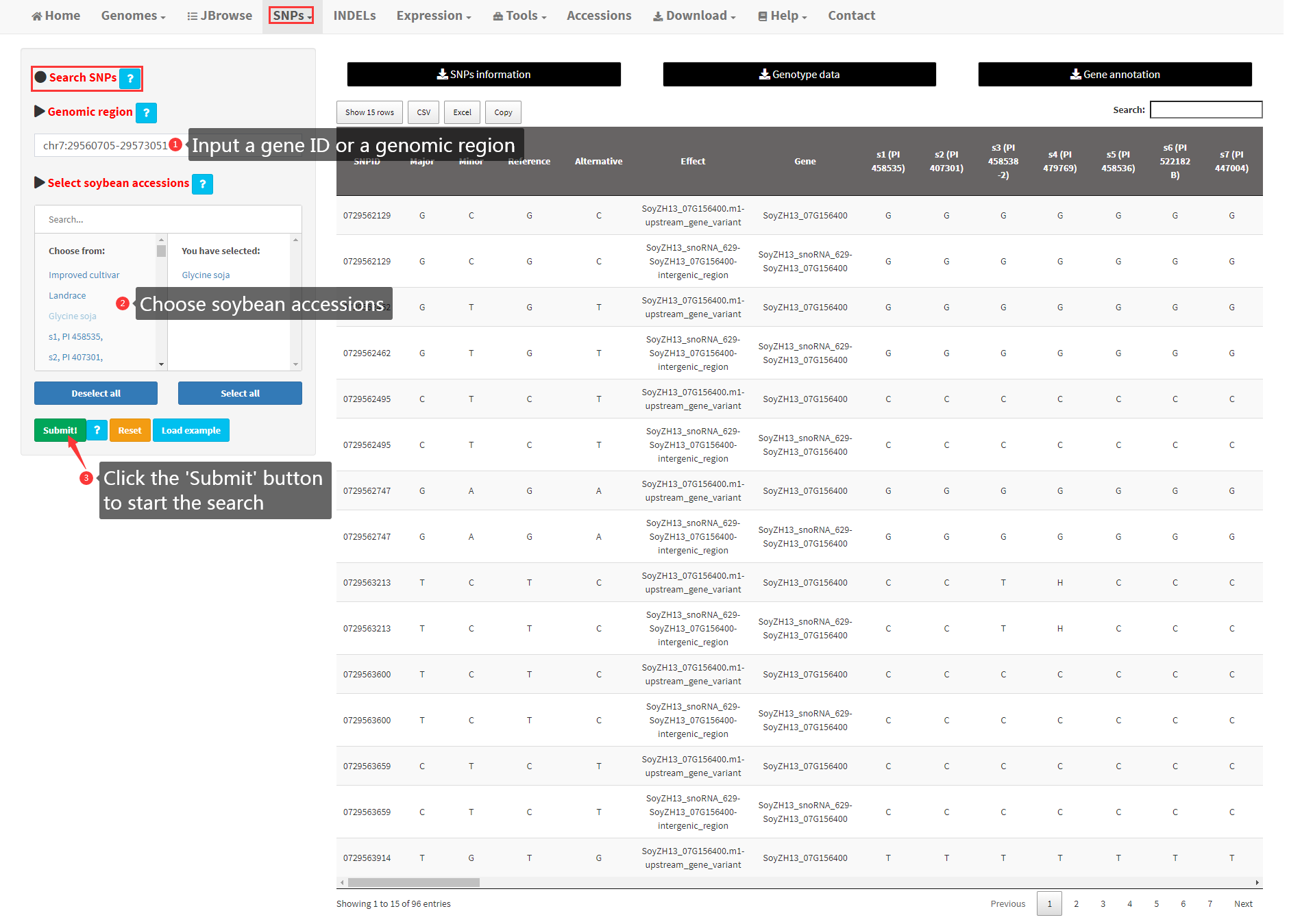
Figure 11. SNPs retrieved in genomic region chr7:29560705-29573051
10. Linkage disequilibrium analysis of SNPs
In this menu, a heatmap can be created to display the linkage disequilibrium between pair-wise SNP sites in a user-input genomic region. Essential steps to conduct linkage disequilibrium analysis is shown in Figure 12. Several options are provided to tune the appearance of the heatmap including figure flipping and colors (Figure 12). Finally, the heatmap can be downloaded in PDF or SVG format.
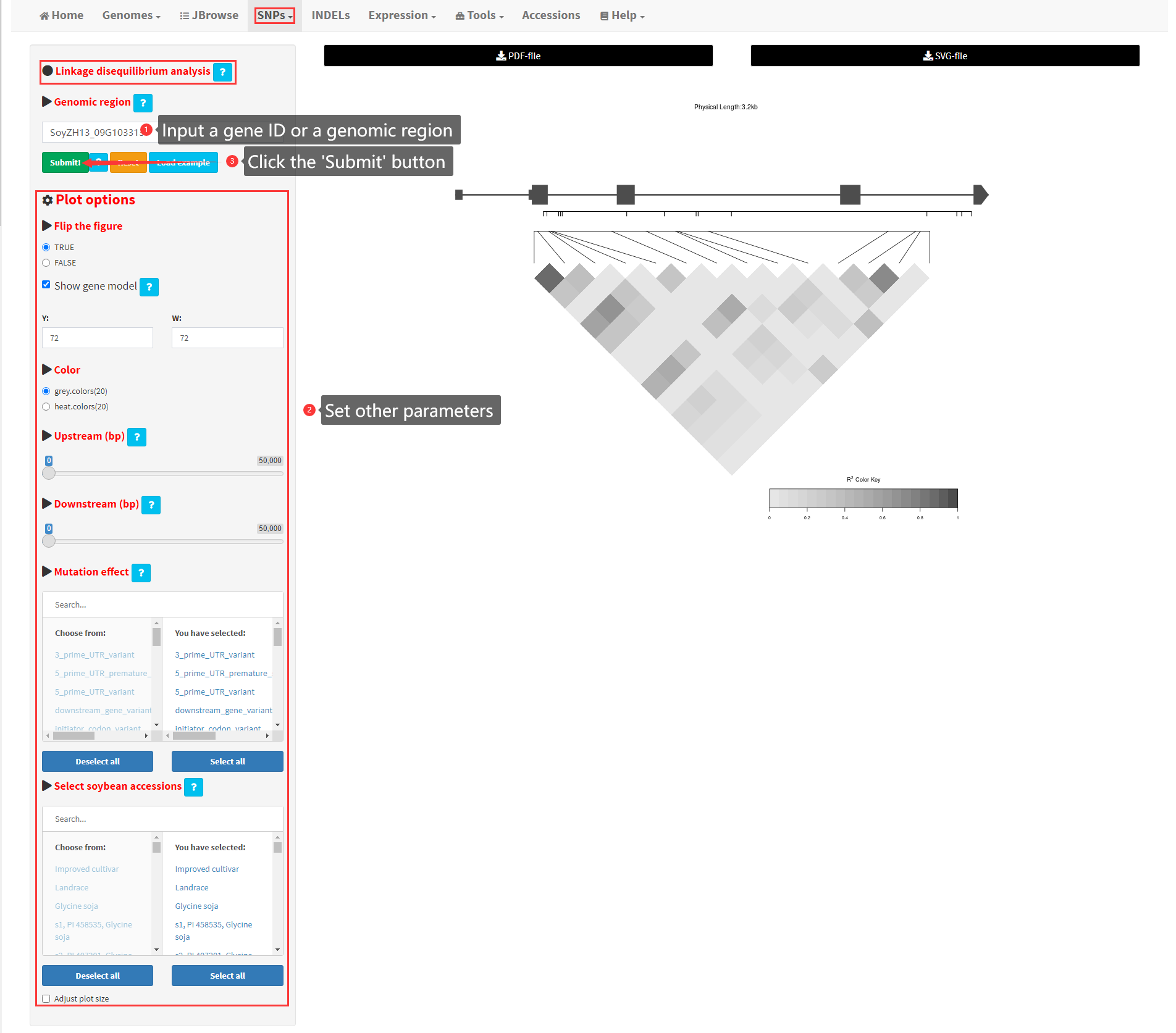
Figure 12. Linkage disequilibrium analysis of SoyZH13_09G103313
11. Nucleotide diversity analysis of SNPs
The ‘‘Diversity” submenu under the “SNPs” menu provides the functionality to calculate nucleotide diversity among groups of soybean accessions in a user-input genomic region. Taking SoyZH13_12G067900 as an example, the results can be adjusted by sequentially setting the widgets “Number of SNPs in each window”, “Ecotypes to calculate diversity”, “Numerator ecotype”, “Denominator ecotype”, “Mutation effect” and “Upstream/Downstream”. After clicking the Submit! button, the results can be visualized in the main panel (Figure 13). The results can also be downloaded as a PDF, SVG or TXT file.
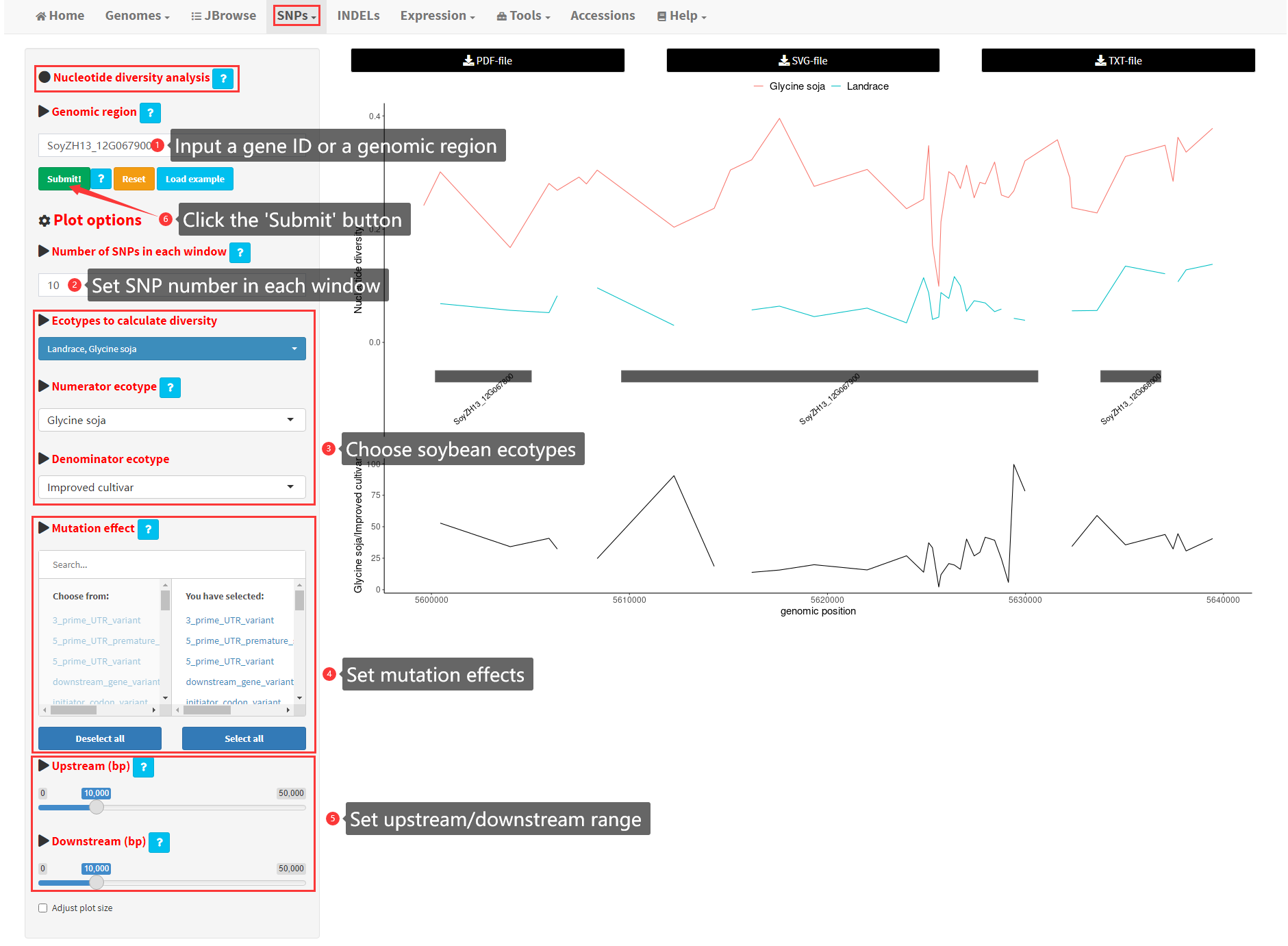
Figure 13. Nucleotide diversity analysis of SoyZH13_12G067900
12. Allele frequency analysis of user-input SNP sites
In this menu, allele frequency of user-input SNP sites across different soybean ecotypes (improved cultivar, landrace, and G. soja) can be calculated and visualized (Figure 14). Several parameters are provided to tune the appearance of the result plot, including the colors used for the major and minor allele and the plot size. After clicking the Submit! button, the results would be visualized in the main panel, which can be exported in PDF or SVG format.
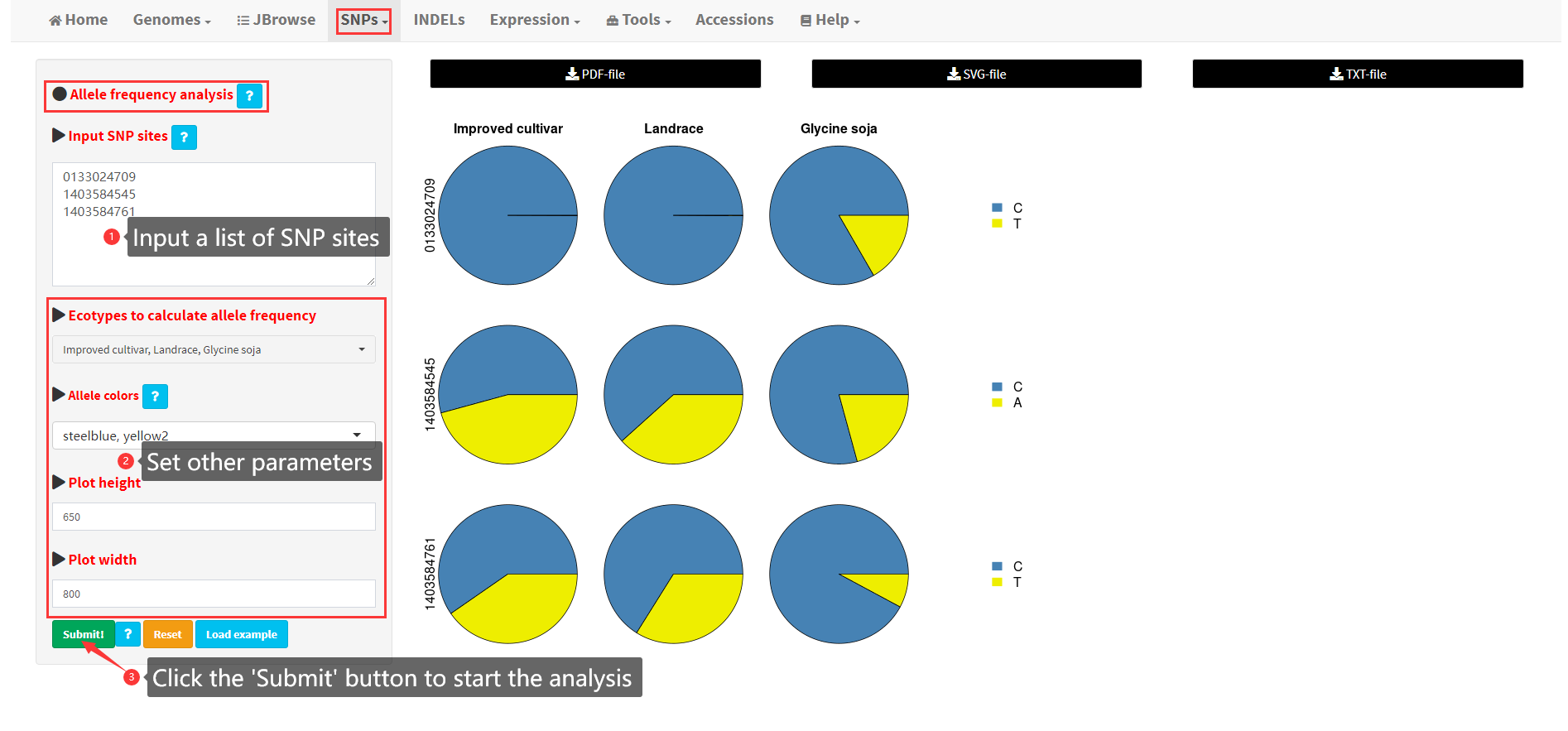
Figure 14. Allele frequency analysis of user-input SNP sites
13. Search and retrieve INDELs
Users can search and retrieve high-quality INDELs among 2898 soybean accessions for any input gene ID or genomic region. Steps to search Indels are shown in Figure 15.
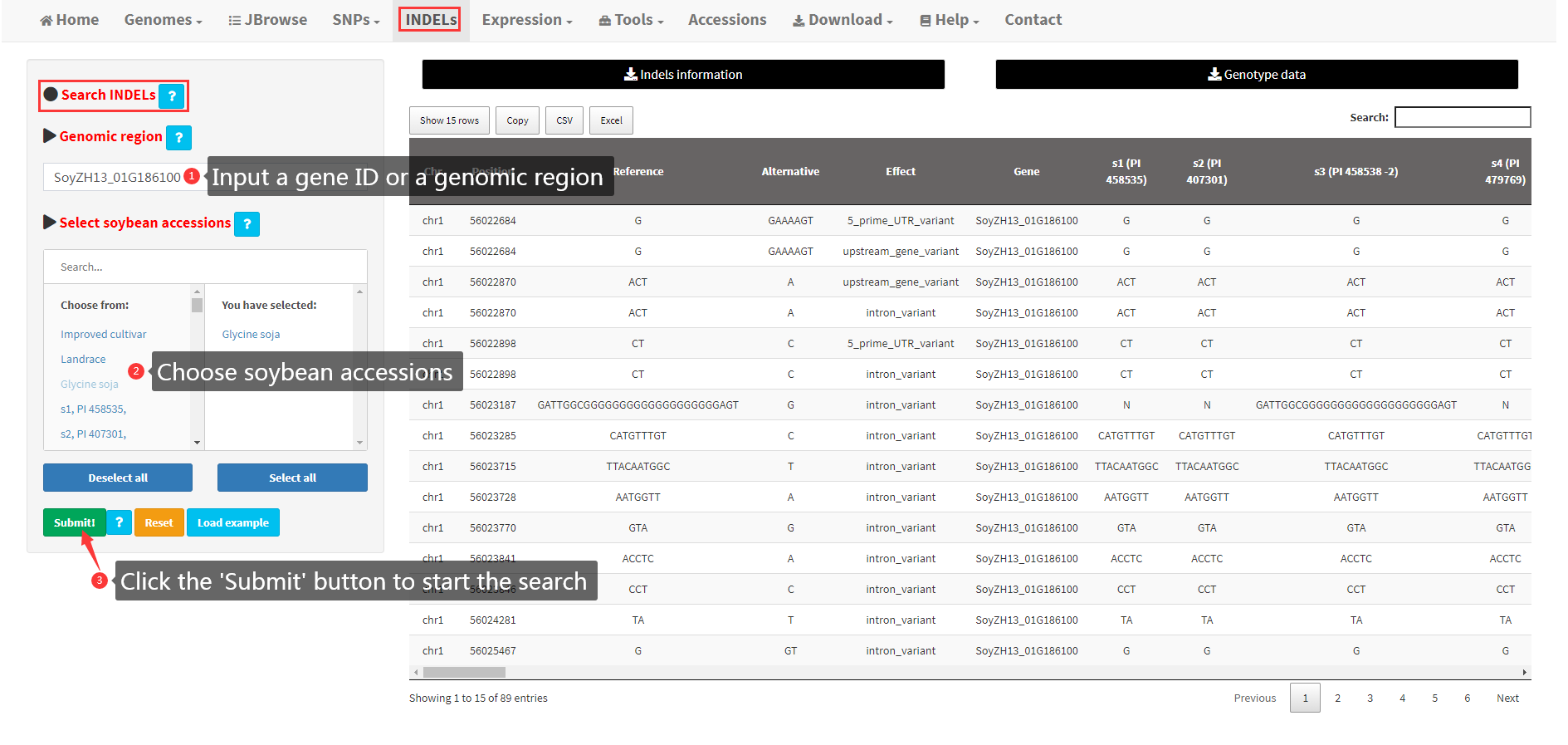
Figure 15. Search and retrieve INDELs located in the genic region of SoyZH13_01G186100
14. Gene expression analysis
The expression levels of protein-coding genes in the genome of Zhonghuang 13 across 27 different tissues/stages were collected in SoybeanGDB. Using the “Gene expression analysis” functionality, the expression level of any user-input gene can be retrieved as a table and visualized as a heatmap (Figure 16). The expression level of the input gene can be downloaded as a csv or excel file. The size of the heatmap can be adjusted using provided widgets in the sidebar panel.
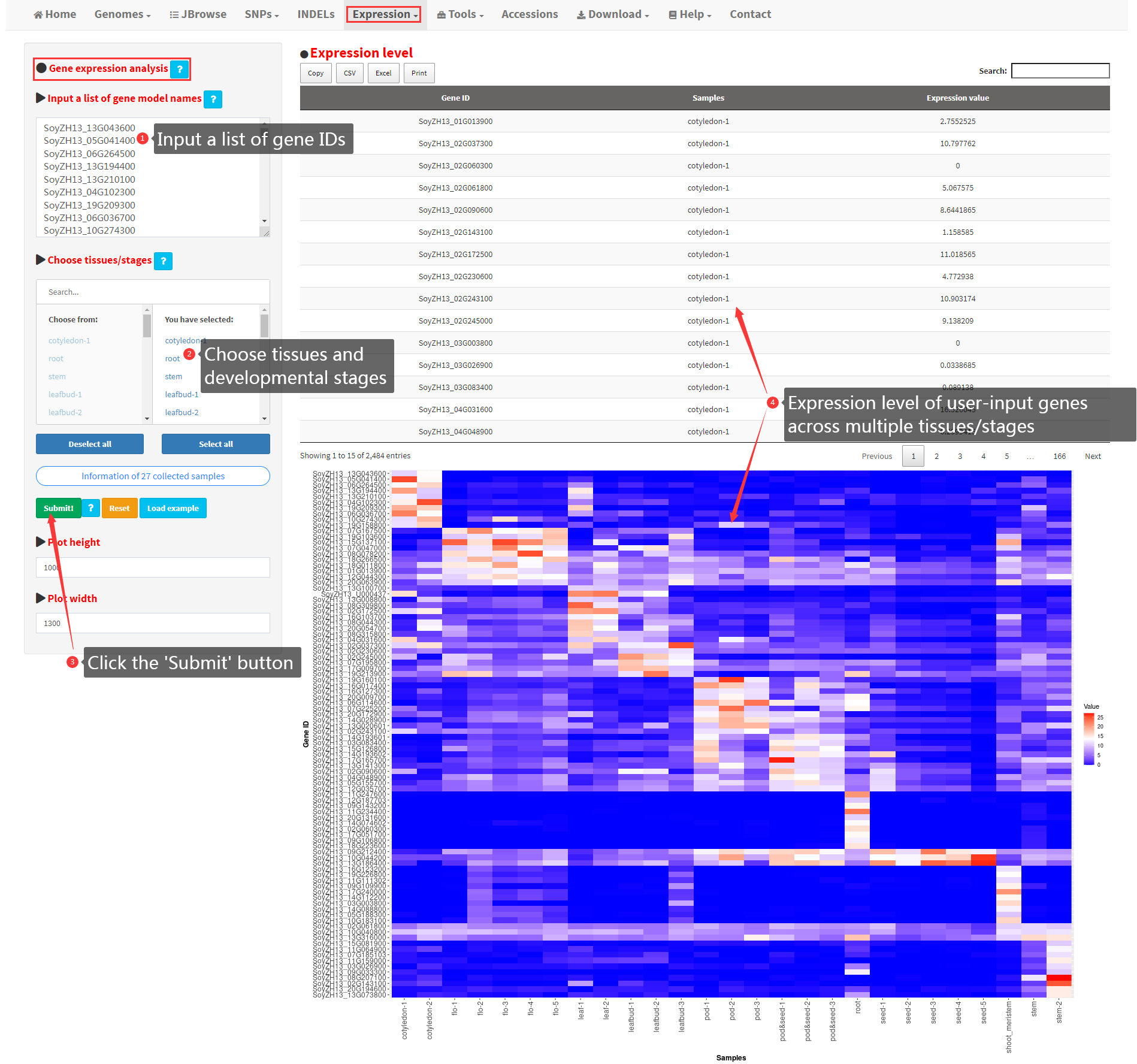
Figure 16. Expression profile of SoyZH13_05G201900
15. Gene co-expression analysis
Based on the expression levels of protein-coding genes in the genome of Zhonghuang 13 across 27 different tissues/stages, we implemented a functionality in SoybeanGDB for users to perform co-expression analysis of input genes. For a user-input gene list, the expression correlation coefficient between the input genes and all expressed genes were calculated and displayed in a table, which can be downloaded as csv or excel files (Figure 17). The expression correlation coefficient can also be visualized as a heatmap. Essential steps to conduct co-expression analysis are shown in Figure 17.
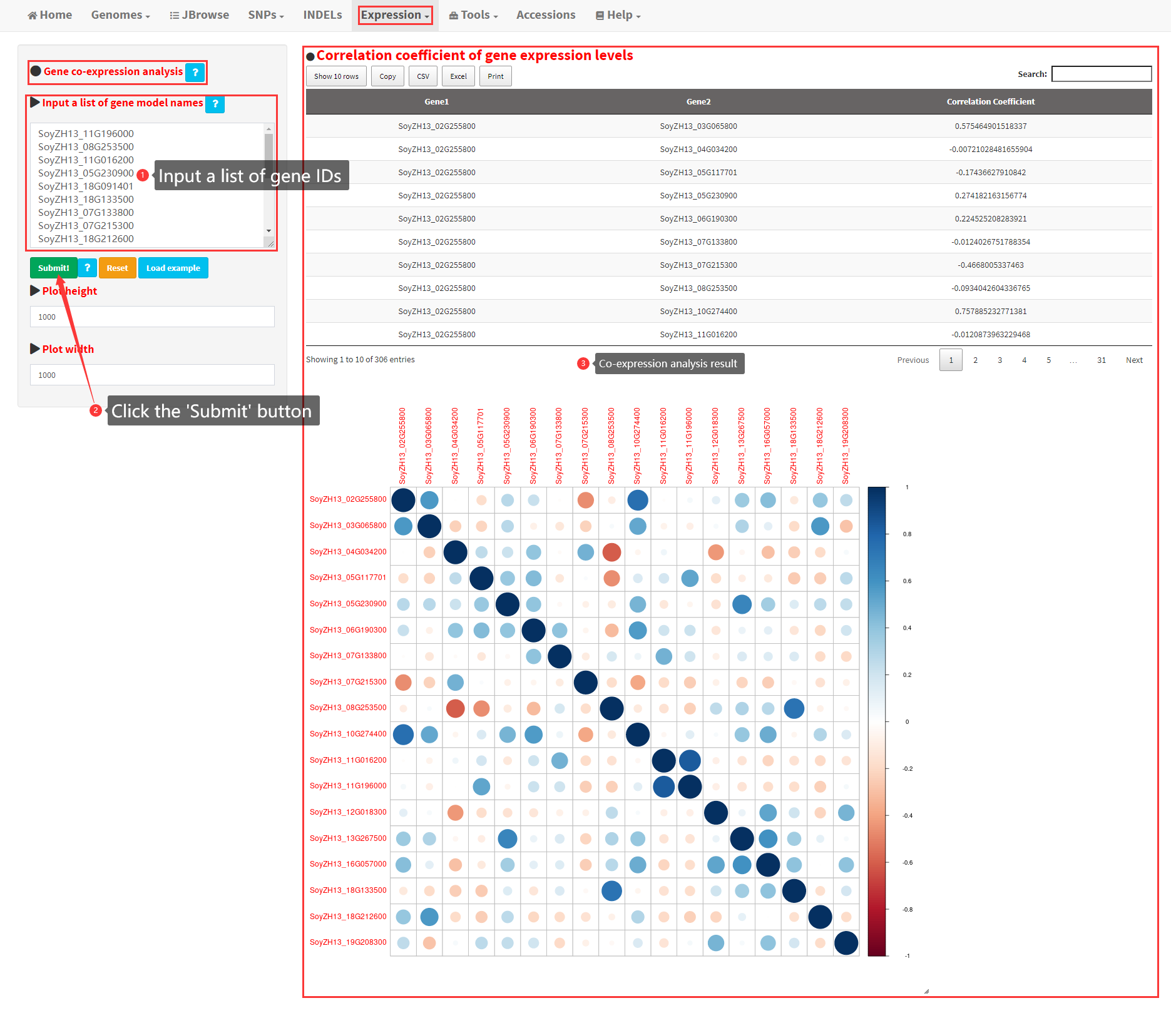
Figure 17. Gene co-expression analysis
16. Search a genome by BLAST
Under this menu, users can search one or multiple soybean genomes by sequence similarity utilizing BLAST (Figure 18). The input sequences must be in fasta format. The genome sequences, gene sequences, CDS sequences, and protein sequences of any one or multiple of the 39 high-quality genomes can be searched by BLAST. After clicking the Submit button, BLAST alignment would be conducted and the results can be viewed in the Output panel (Figure 19).
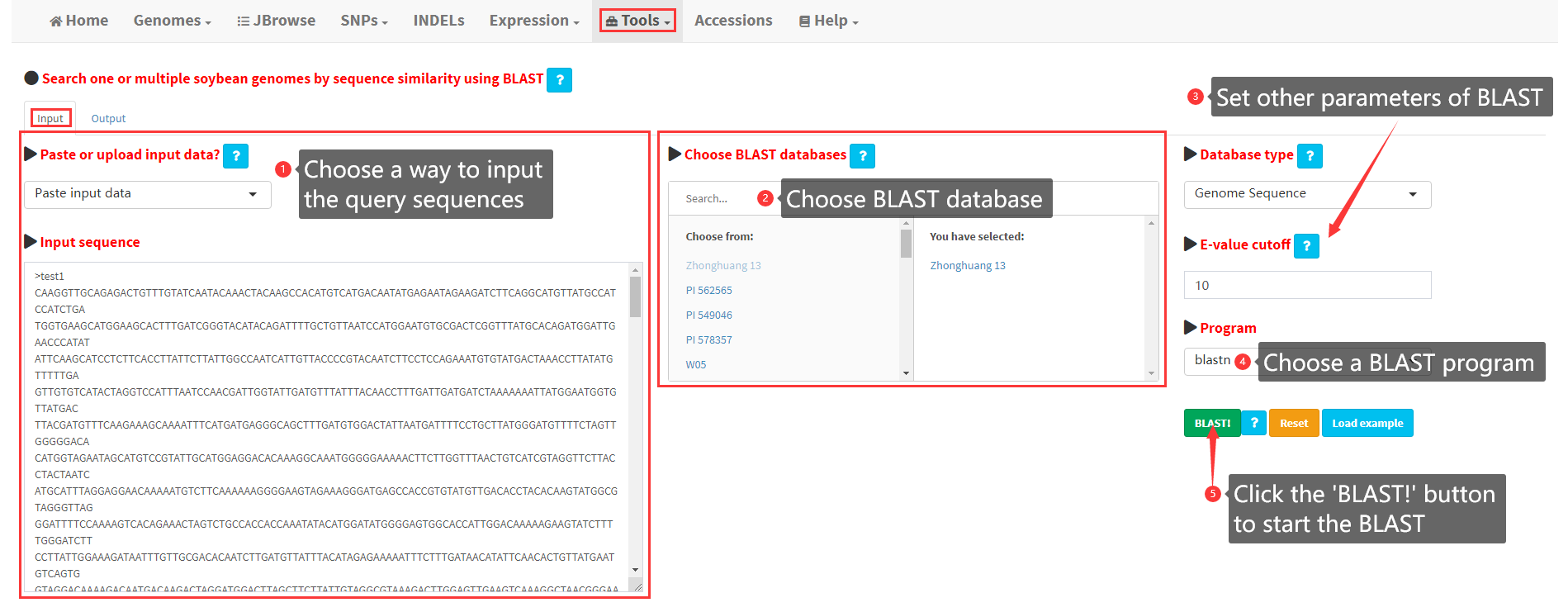
Figure 18. Steps to perform BLAST
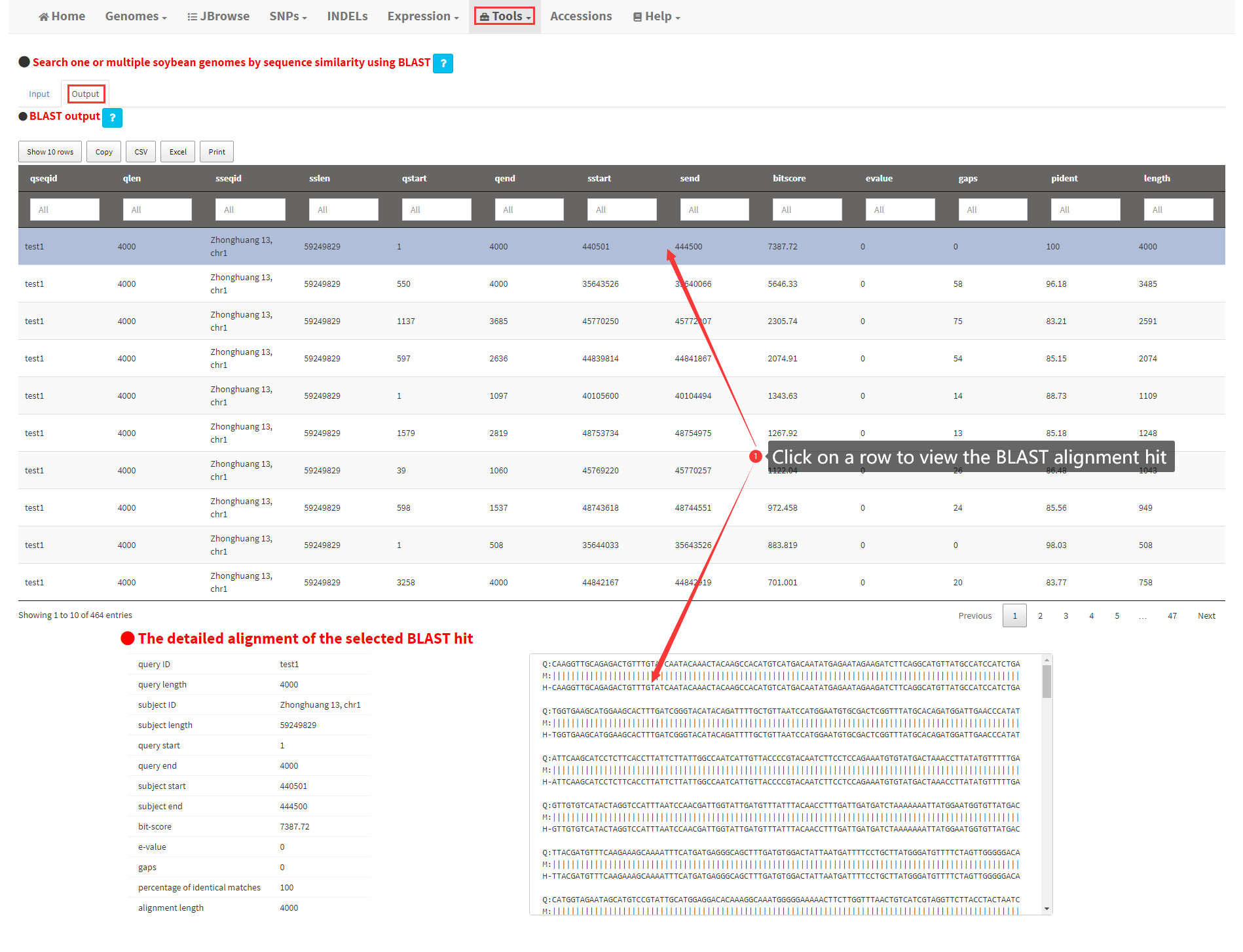
Figure 19. BLAST result viewed in the output panel
17. Design primers for a user-input genomic region or gene locus
Using this functionality, users can design primers for any input genomic region or gene locus in the genome of Zhonghuang 13, targeting SNPs and Indels in this region (Figure 20). Primer3 was utilized to design the primers. Many options of Primer3 are implemented as graphical interface for users to set appropriate parameters for primer designing. Five best candidate primers would be displayed in a result table, with each column representing diverse features of the designed primers. The detailed information of the best primers is displayed below the table, including the template sequence, the primers sequence and the SNPs/INDELs in the input region.
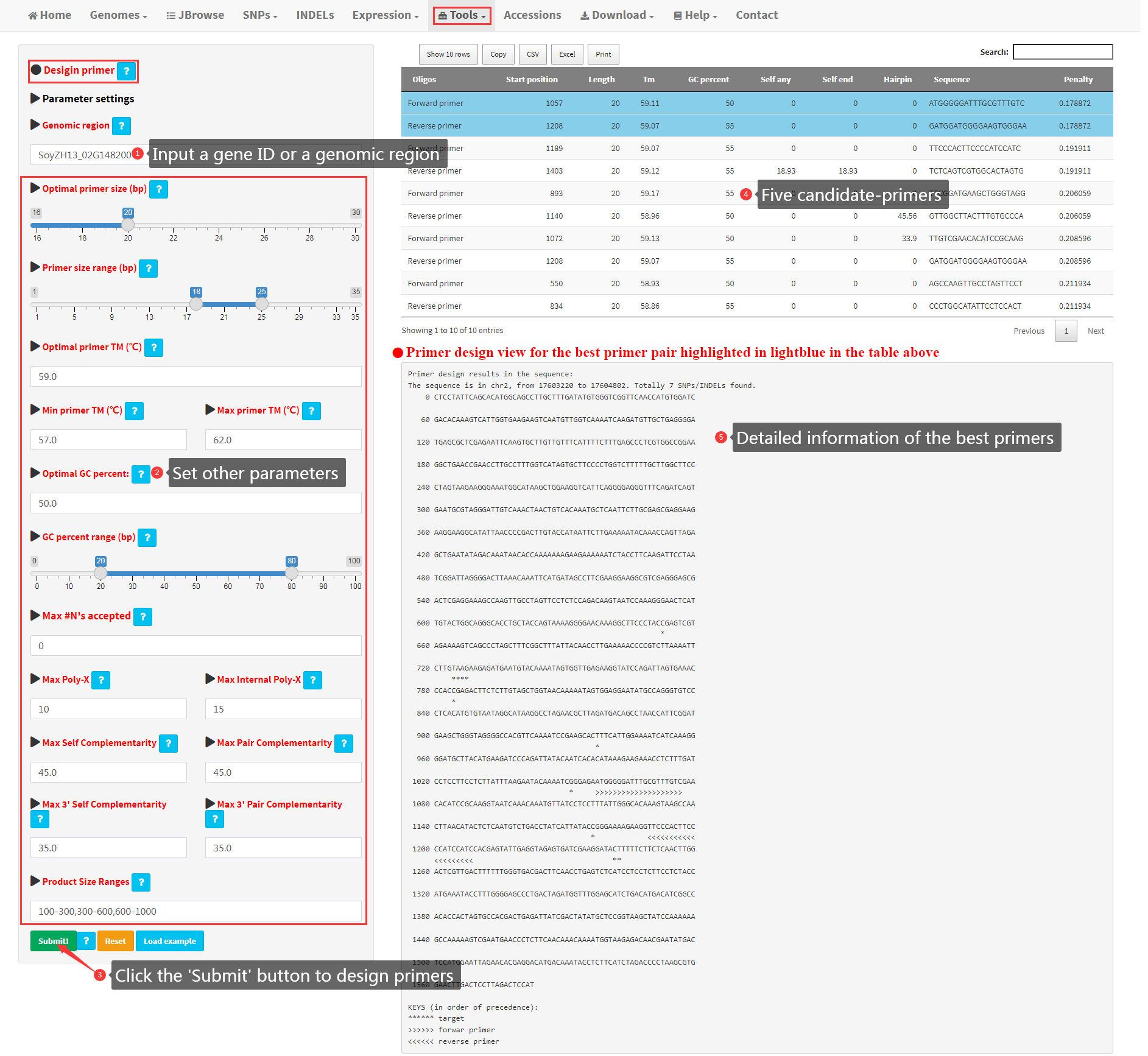
Figure 20. Design primers for a user-input genomic region
18. Search orthologous gene groups among 39 genomes
With this functionality, users can search orthologous groups among 39 soybeans genomes by inputting a single gene ID of any of the 39 genomes (Figure 21). Then, orthologs of the input gene in other genomes would be displayed in the main panel after clicking the Submit! button.
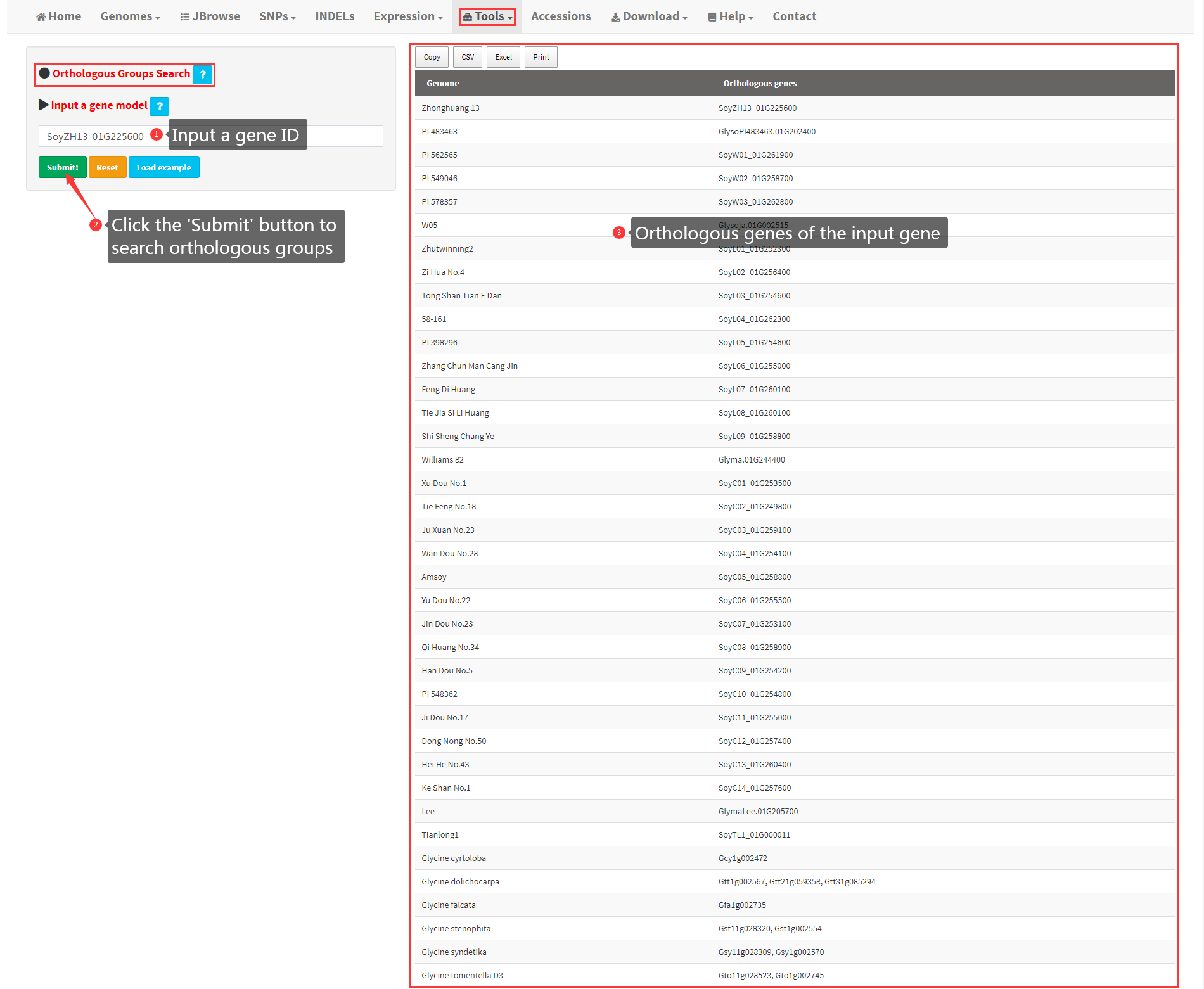
Figure 21. Orthologous genes search
19. GO Annotation of a user-input gene list
This functionality is used to perform GO annotation of a user-input gene list from any of the 39 soybean genomes. Steps to conduct GO annotation are shown in Figure 22. The full annotation result is displayed as a table, which can be downloaded. The top 30 GO terms with the largest number of genes are displayed as a bar plot (Figure 22).
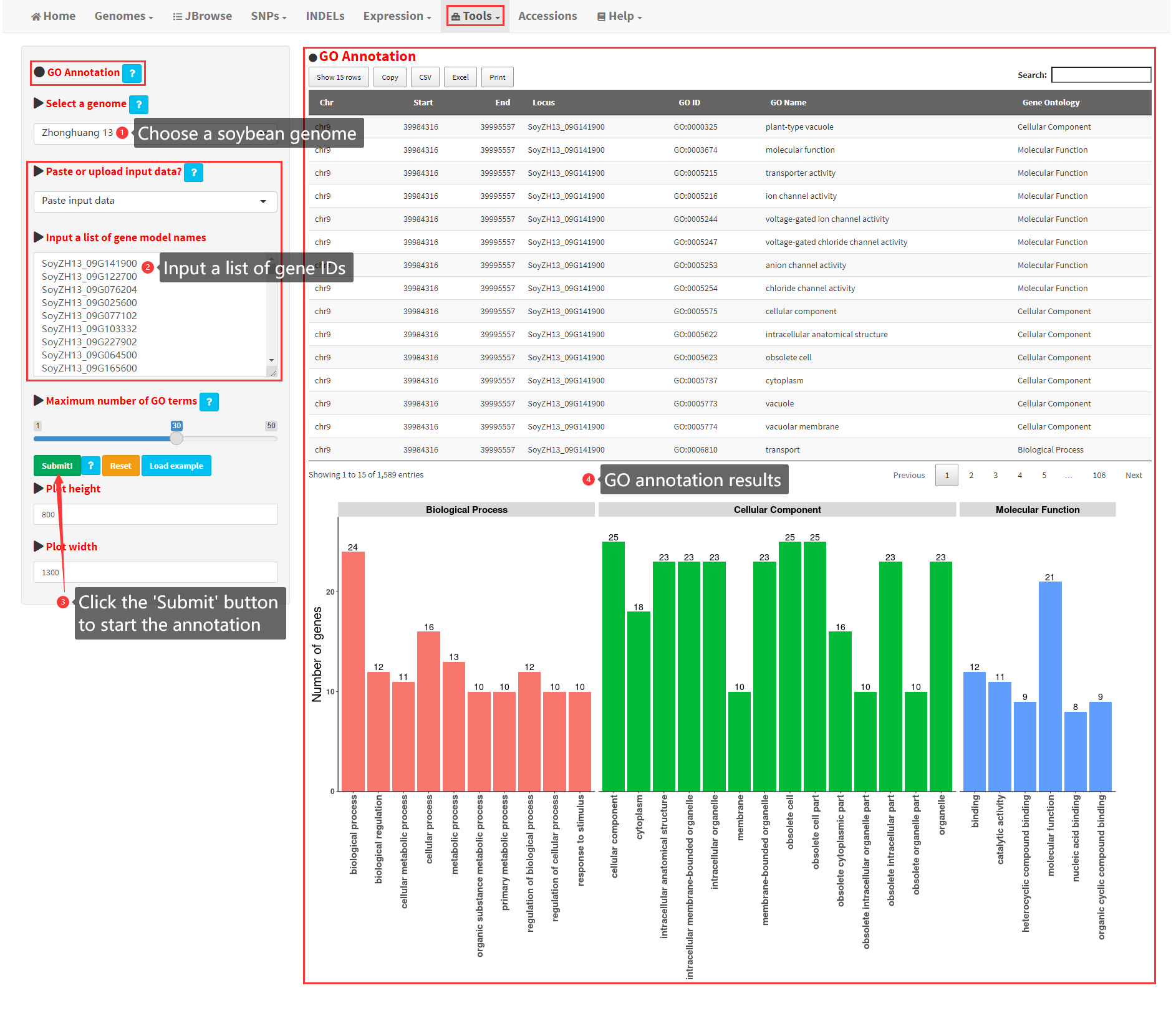
Figure 22. GO Annotation of user-input genes
20. GO enrichment analysis of a user-input gene list
This functionality is used to perform GO enrichment analysis of a user-input gene list from any of the 39 soybean genomes. Steps to conduct GO enrichment analysis are shown in Figure 23. Enrichment analysis for each GO category (Molecular Function, Biological Process, Cellular Component) was conducted separately. For each category, the full enrichment result is displayed as a table, and the significant enrichment result is displayed in two figures. The enrichment result can be further filtered by other parameters, including adjusted P value, Q value, etc. (Figure 23).
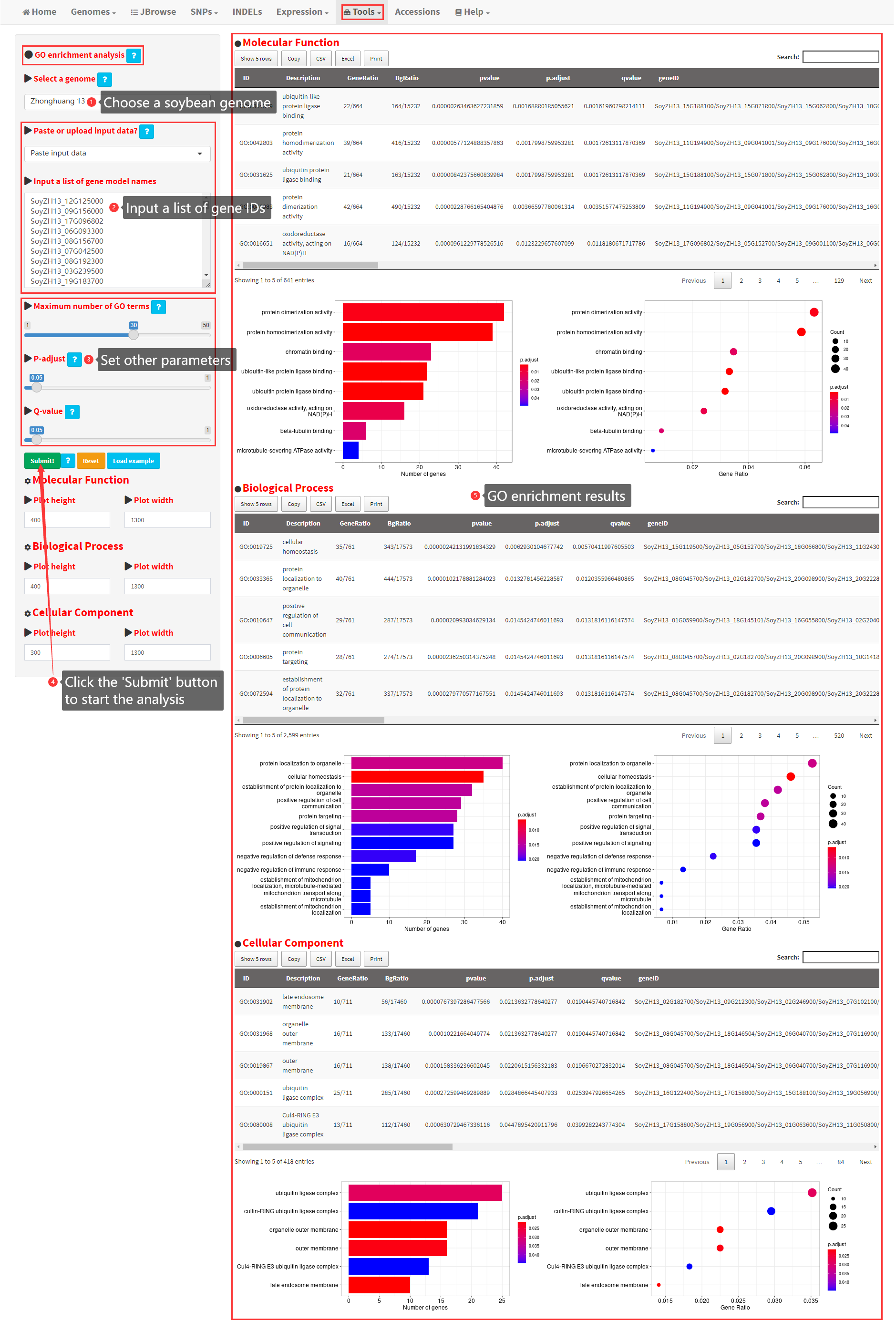
Figure 23. GO Enrichment analysis of user-input genes
21. KEGG Annotation of a user-input gene list
This functionality is used to perform KEGG pathway annotation of a user-input gene list from any of the 39 soybean genomes. Steps to conduct KEGG annotation are shown in Figure 24. The full annotation result is displayed as a table, which can be downloaded. The top 30 largest KEGG pathways are displayed as a bar plot (Figure 24).
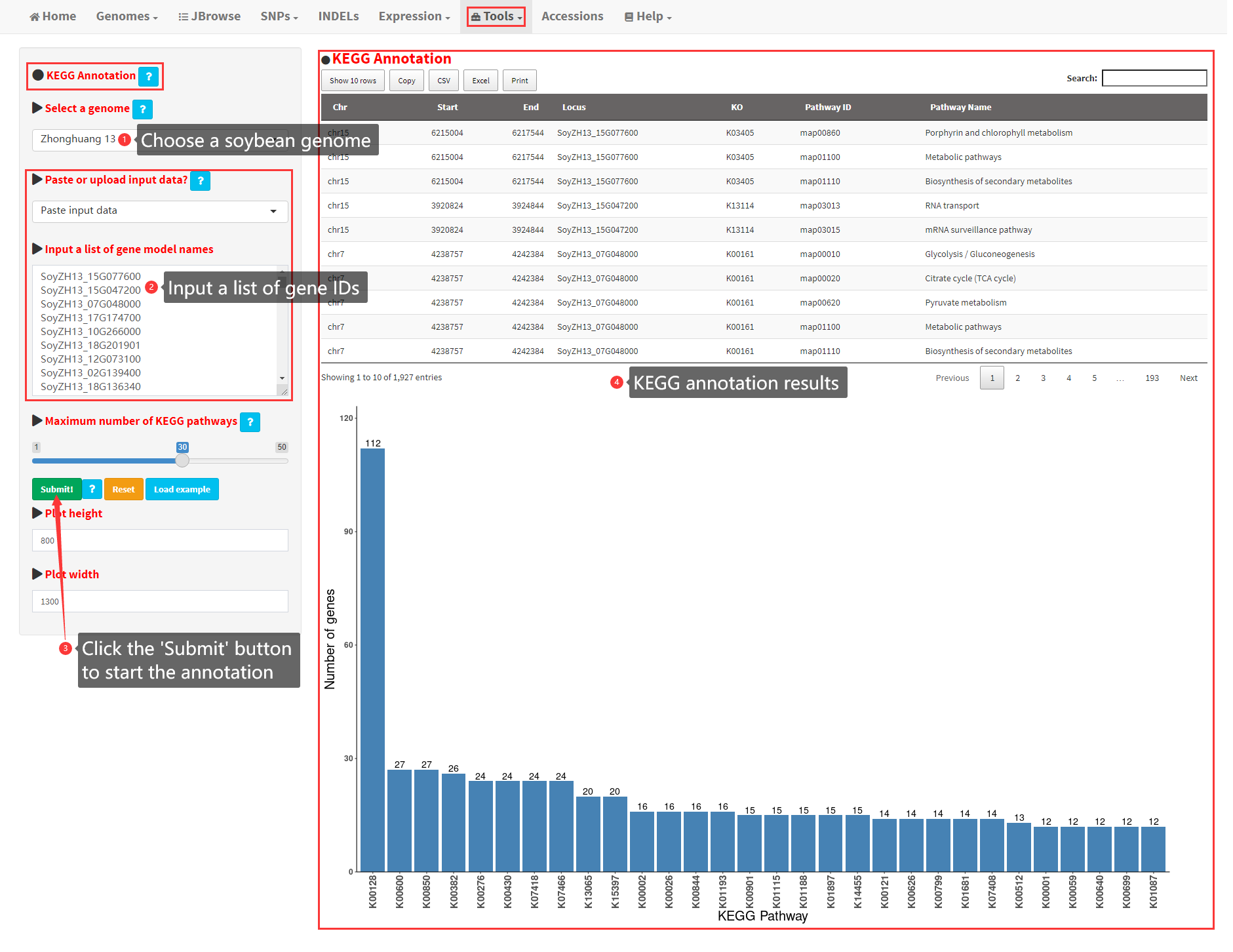
Figure 24. KEGG Annotation of user-input genes
22. KEGG enrichment analysis of a user-input gene list
This functionality is used to perform KEGG enrichment analysis of a user-input gene list from any of the 39 soybean genomes. Steps to conduct KEGG enrichment analysis are shown in Figure 25. The full enrichment result is displayed as a table, and the significant enrichment result is displayed as a figure. The enrichment result can be further filtered by other parameters, including adjusted P value, Q value, etc. (Figure 25).
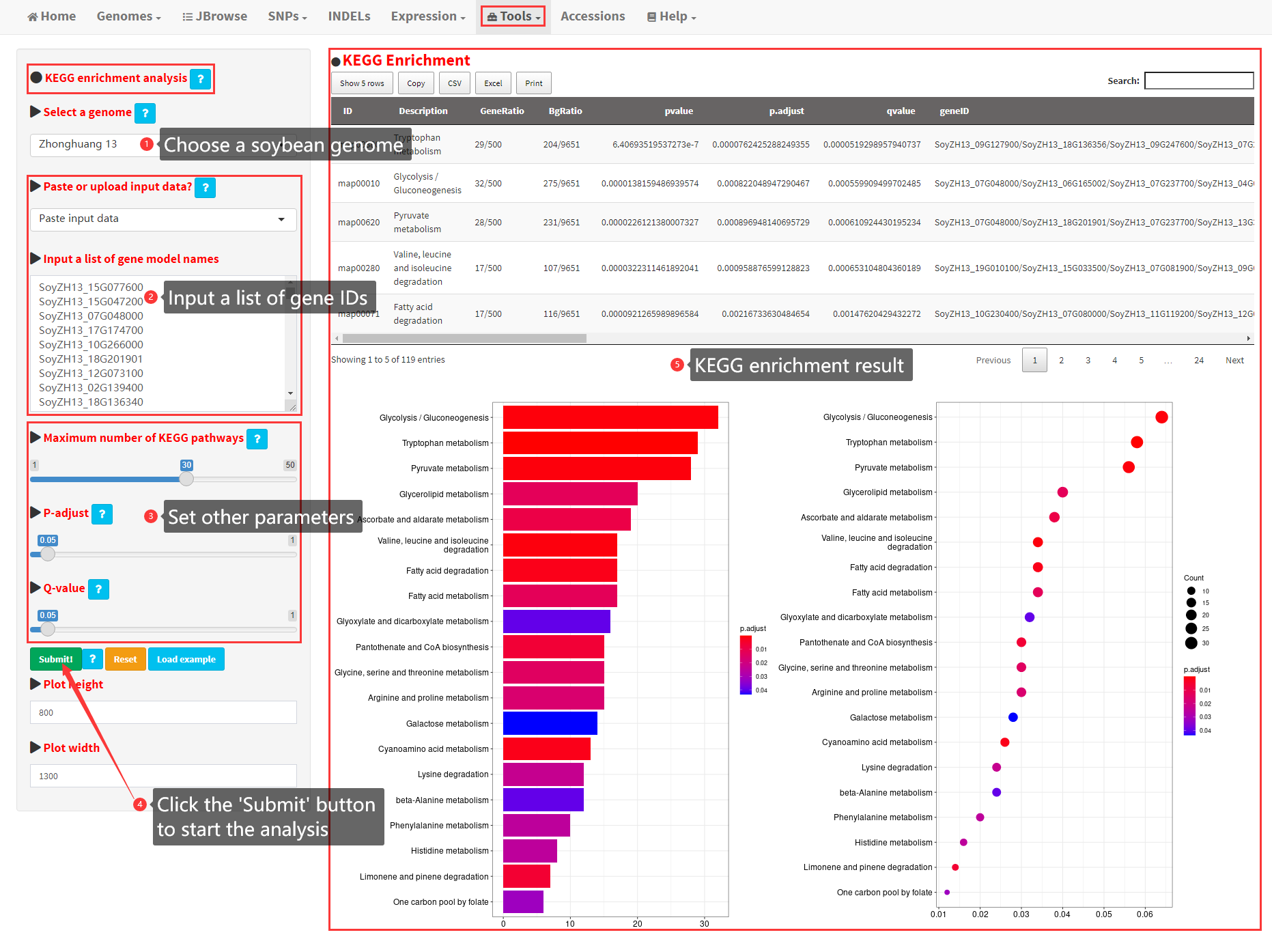
Figure 25. KEGG Enrichment analysis
23. Information of 2898 soybean accessions
A total of 2898 soybean accessions were collected in SoybeanGDB. The detailed information of all 2898 accessions is displayed in the “Accessions” submenu under the “Help” menu of SoybeanGDB, which can be downloaded as a csv or excel file. In the sidebar panel of the “Accessions” submenu, a widget is provided for users to select one or multiple accessions to view the detailed information in the main panel (Figure 26).
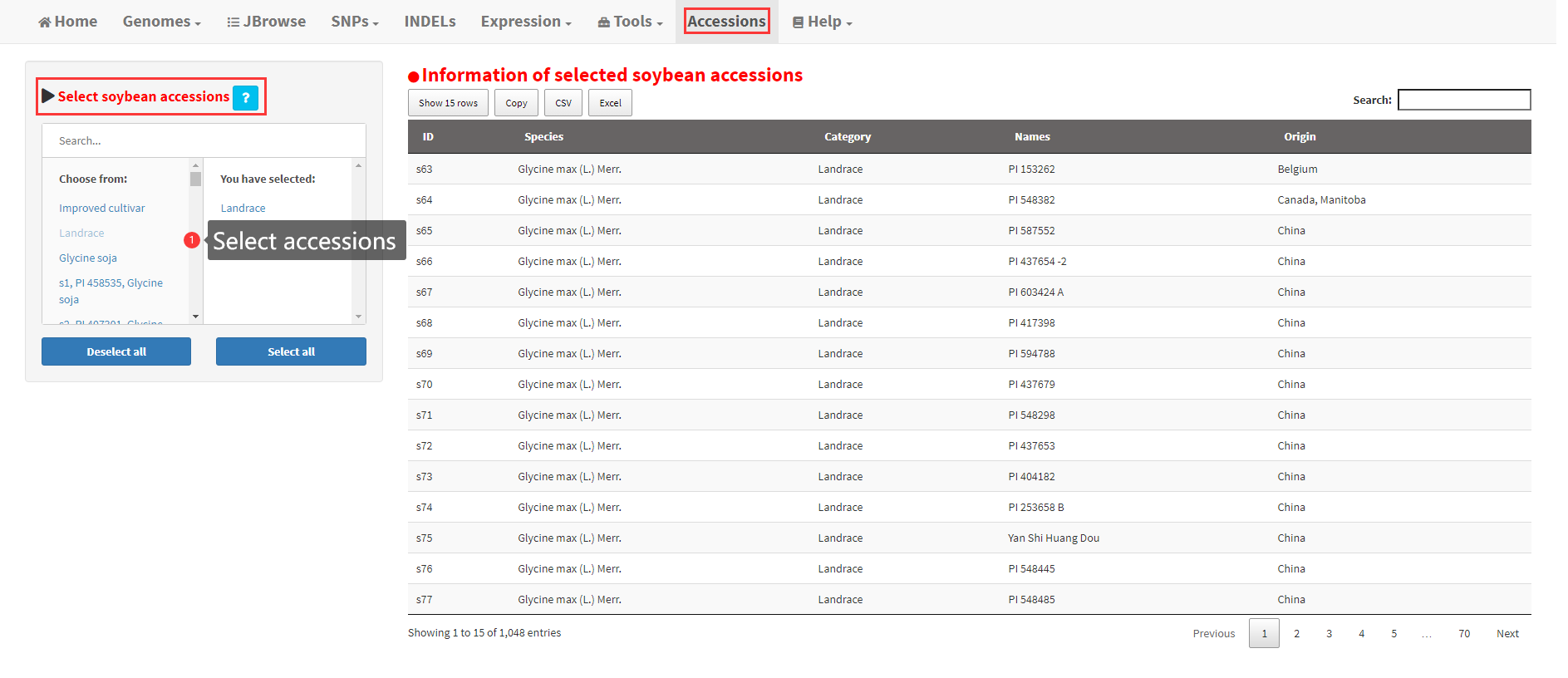
Figure 26. Retrieve information of soybean accessions
2023.03.07
- New expression dataset added: Genic C-Methylation in Soybean Is Associated with Gene Paralogs Relocated to Transposable Element-Rich Pericentromeres
2023.03.07
- New expression dataset added: RNA-Seq Atlas of Glycine max: A guide to the soybean transcriptome
2023.02.21
- New SNPs dataset added: Genetic variation among 481 diverse soybean accessions, inferred from genomic re-sequencing
2022.10.21
- A new page: “Literature” added
2022.05.21
- Hwangkeum genome added: Genome assembly of the popular Korean soybean cultivar Hwangkeum
2022.04.13
- Six Perennial Glycine genomes added: Phylogenomics of the genus Glycine sheds light on polyploid evolution and life-strategy transition
2022.01.28
Contact Us
- If you have any problem, comments or suggestions about our database, please contact us.
- e-mail: yaowen at henau.edu.cn and haoran5695 at qq.com
- All comments and suggestions on this database will be highly appreciated.
Literature of Soybean
2023
- 广州大学教授孔凡江团队与合作者关于大豆研究的两项新成果,有望为大豆育种提供重要的基因资源
孔凡江团队与刘斌团队合作,研究揭示了大豆Gmeid1蛋白作为连接光信号和生物钟晚间复合物的桥梁,参与调控大豆开花抑制因子E1基因表达。研究团队通过转录组测序对大豆基因组的节律表达进行分析,筛到一个与开花核心基因E1表达模式完全相反的基因——Gmeid1。 - 中国科学院田志喜团队联合北京基因组研究所章张、宋述慧团队,开发了大豆多维组学深度整合数据库SoyOmics
SoyOmics数据库全面整合分析了大豆相关的多维组学数据。数据库目前收录了27个大豆品系的从头组装基因组数据,并对相应基因组信息进行了全面的基因组注释。该数据库以高质量的ZH13作为参考基因组,对2898份材料的全基因组测序数据进行了全基因组序列变异检测,鉴定到约3800万条SNP/INDEL变异数据,同时为每个变异位点提供多层次注释信息。除序列变异外,数据库提供了来自大豆泛基因组分析的约55万条结构变异数据以及基于结构变异构建的图泛基因组。数据库收录了来自ZH13和Williams82两个基因组27个组织时期的表达数据以及其他26个品系9个组织时期的表达数据,并展示了不同品系间同源基因的差异表达。数据库针对115个表型多年多点测定的约2.7万条表型记录进行了本体注释和归类,并将表型数据与变异数据进行关联。除以上组学数据外,数据库同时提供了部分种质资源的甲基化测序数据以及Soy40K大豆芯片数据。该数据库从基因组、变异组、转录组、表型组等不同层面整合了大豆相关数据集,实现了不同层次组学数据的交互查询和联合比较分析。 - 中国科学院遗传与发育生物学研究所的研究团队首次构建了大豆的泛三维基因组
科研人员根据基因组重头组装的27份大豆种质材料,利用高通量染色质构象捕获技术,获得高质量的三维基因组数据,从A/B区室和拓扑关联结构域层面解析了各个大豆基因组三维构象的保守性和特异性,同时,利用泛基因组分析鉴定的存在与缺失变异与拓扑关联结构域联合分析,首次解析了转座元件在种质特异性的拓扑关联结构域边界形成中的作用,重塑了大豆的三维基因组。
2022
- 中国农业科学院作物科学研究所发掘了大豆不同进化阶段受到选择的候选基因
中国农业科学院作物科学研究所大豆优异基因资源发掘与创新利用团队联合国内外科研机构,通过解析大豆地理扩张与育种的全基因组特征,提出大豆进化路线,发掘了大豆不同进化阶段受到选择的候选基因,从中克隆了一个重要的开花基因GmSPA3c。 - 高产、抗SMV春播鲜食大豆‘苏奎3号’的选育及栽培技术
为了筛选出高产、优质、抗性强、生育期适中,适宜在江苏省推广的鲜食大豆,以‘辽鲜2号’和‘苏早1号’为试验材料进行杂交,其杂交后代经过多年多点的试验鉴定,最终选育出高产、抗大豆花叶病毒(SMV)鲜食春大豆新品种‘苏奎3号’。2016—2017年参加江苏省区试,‘苏奎3号’年平均鲜荚产量较对照品种‘台湾292’增产18.92%。2018年参加江苏省春播鲜食大豆生产试验,‘苏奎3号’平均每公顷产鲜荚较对照增产16.58%。2019年6月通过江苏省农作物品种审定委员会审定(苏审豆20190006)。‘苏奎3号’具有高产以及抗SMV等特点,适宜在江苏各地作春播鲜食大豆种植使用。本文对该品种的特征特性、高产栽培技术作一阐述,供相关研究单位及农技推广部门参考。 - 东北农业大学刘珊珊教授团队和张淑珍教授团队发表降胆固醇特殊健康用途“功能大豆新品种创制”相关论文
该研究利用六个7S与11S球蛋白亚基组成各异的大豆为原料,采用碱溶酸沉法制备大豆分离蛋白,设置不同处理因素[不同基因型(SPI-1~6)(300 mg/kg/day)及菲诺贝特(30 mg/kg/day)],灌喂随机分成九组的54只高胆固醇模型鼠。28天后,采血测定血脂、采集肝脏及内脏脂肪,观察其病理学变化,测定肝脏总胆固醇(TC)、甘油三脂(TG)含量及胆固醇代谢相关关键基因的mRNA表达量(图2)。结果表明,大豆基因型影响其SPI的降胆固醇功效,不同7S与11S球蛋白亚基组成SPI的降胆固醇功效差异显著,7S-(α′+α)双缺失型“东农豆360”制备的蛋白粉“SPI-2”可以显著提高高胆固醇模型鼠血清的HDL-C水平。不同亚基组成SPI降胆固醇的分子机理各不相同,qRT-PCR结果显示,灌喂28天“东农豆358”制备的保健蛋白粉“SPI-3”可使高胆固醇模型鼠肝脏中的Cyp7a1基因显著上调表达。 - 中科院遗传发育所在Plant Biotechnology Journal 发表研究论文,克隆调控大豆籽粒大小和品质的重要基因
中国科学院遗传与发育生物学研究所植物细胞与染色体工程国家重点实验室田志喜研究组通过对1800多份大豆种质资源籽粒厚度性状的全基因组关联分析,在5号染色体上鉴定到了一个与粒厚相关的稳定信号区间,并且该区间与已报道的多个产量、油含量和蛋白含量候选QTL区间重叠。进一步结合多组学数据分析,确定了一个控制大豆籽粒厚度和大小的基因GmST05(Seed Thickness 05)。GmST05编码一个PEBP家族的蛋白,是拟南芥AtMFT的直系同源基因。单倍型分析发现,GmST05启动子区的自然变异位点显著影响其表达量,GmST05HapⅠ表达水平明显高于GmST05HapIⅠ,是决定大豆种子大小的关键因素。转基因实验表明GmST05正调控大豆籽粒大小。进一步的研究发现GmST05可能通过调控GmSWEET10a的转录,影响籽粒油含量和蛋白含量。有意思的是,GmST05的等位基因变异存在明显的地理分布差异,即高纬度地区的大豆种质普遍拥有GmST05HapⅠ单倍型,而低纬度地区大豆种质普遍拥有GmST05HapⅡ单倍型。群体遗传多样性分析表明,GmST05的等位基因变异在大豆进化过程中正在经历着人工选择,但尚未被固定。这为挖掘和利用GmST05优良等位变异提高大豆产量提供了重要基因资源和理论基础。 - 中科院遗传发育所在Nature Communications发表研究论文,揭示大豆分枝数的调控机制
利用全基因组关联分析挖掘到大豆分枝数主效控制基因Dt2。遗传分析发现,Dt2负调控大豆分枝数,CRISPR/Cas9基因敲除株系分枝数明显增多,小区产量明显提高;过表达株系分枝数显著降低,小区产量明显下降。进一步,通过酵母双杂交、双分子荧光互补和免疫共沉淀实验发现,Dt2、GmAgl22和GmSoc1a蛋白可以两两互作。转录组、生物化学和遗传学分析发现,Dt2能够直接结合到GmAp1a和GmAp1d的启动子上,正调控GmAp1a和GmAp1d的转录,同时GmSoc1a和GmAgl22能促进Dt2对GmAp1a和GmAp1d的转录。由此推演出Dt2调控大豆分枝的分子调控模型:Dt2可结合到GmAp1a和GmAp1d的启动子正调控其转录,GmAgl22和GmSoc1a与Dt2的互作可增强了Dt2的转录激活效应。GmAp1a和GmAp1d抑制大豆分枝。 - 【Nature Com】大豆突变一个基因,含油/产量上升,蛋白质量下降
该研究证明了 CCT 基因POWR1是蛋白质/油含量的大效应的QTLl(large-effect QTL )的基础。研究表明POWR1中的TE 插入会截断其 CCT 结构域并显着增加种子油含量、重量和产量,同时降低蛋白质含量。POWR1多效性可能通过调节种子养分转运和脂质代谢基因来控制这些性状。 - 中科院东北地理所等合作在生物学著名期刊(IF=10.372)上发表研究论文,成功克隆新型调控大豆营养生长期长度的新基因QNE1
本研究揭示了与光周期E1通路迥异的基因调控通路,进一步丰富了大豆开花调控多样性,同时为通过分子设计育种培育广适性大豆栽培品种奠定了重要的基础。 - 【Science】大豆中过表达三个基因,田间最高增产33%
综上所述,At VDE、At PsbS 和At的过表达在为期 2 年的田间试验中,大豆中的 ZEP 促进了 NPQ 弛豫的加速,同时在波动光下光合效率也随之提高。通过加速 NPQ 放松,在 2020 年观察到五个独立事件的平均种子产量增加了 24.5%。 - 中科院东北地理所在Plant Physiolgoy杂志上发表研究论文,在大豆光周期现象分子调控机制研究上取得重要进展
大豆是典型的光周期敏感短日照作物,大豆生育期(开花期与成熟期)及株型都严格受光周期调控。短日照条件下,大豆的生育期短,植株矮小,长日条件下大豆的生育期延长,植株茎节增多,植株高大。大豆光周期敏感性使单个大豆品种适宜种植范围狭窄,造成了许多优良品种的推广范围小,有“宝”难用,影响了我国大豆的稳产与高产。中国科学院东北地理与农业生态研究所大豆分子设计育种课题组于2012年首次在大豆中成功克隆了对光周期反应及生育期(开花期及成熟期)贡献最大的基因E1(PNAS, Xia et al., 2012)。E1编码豆科植物特异性转录因子,是大豆光周期响应的核心调控因子,阐明其介导开花途径的分子机制尤为重要。然而,目前有关E1所调控的下游信号通路在很大程度上仍是未知的。 - 华南农业大学在著名期刊(即时影响因子8.48)发表研究论文,揭示不同测序方法对大豆耐涝根际微生物机制的影响
该项研究利用Illumina Miseq、LoopSeq和PacBio三代全长测序,研究了涝害对中性和酸性土壤大豆根际细菌群落结构的影响。三种测序方法的结果都表明,涝害胁迫显著改变了两种土壤的根际微生物群落结构,并增加了固氮微生物(Geobacter)的丰度。此外,与16S(Illumina Miseq)测序相比,全长(LoopSeq和PacBio)16S测序具有更高的分辨率。核心微生物和网络模块化分析显示,三种测序方法都发现涝害增加了与氮循环相关微生物的相对丰度,而LoopSeq和PacBio测序方法还发现涝害增加了与磷循环相关微生物的相对丰度。核心微生物和网络模块化分析进一步揭示,不同物种的富集可能在维持细菌群落结构和生态功能的稳定性方面起着核心作用。 - The Crop Journal | 中国农科院作科所和基因组所揭示大豆驯化改良过程中有害突变模式
作者在已发表的2214份大豆(221个野生大豆、1132个地方品种和861个栽培品种)全基因组重测序数据中共鉴定到115,272个有害SNP。发现有害等位基因数目在驯化过程(野生大豆到地方品种)显著增加,而在改良过程(地方品种到选育品种)显著减少(图1)。 位于selective sweep区的基因比其他基因组区段包含更少的有害突变(图1)。野生大豆从我国南部向中部和北部扩散过程中,有害突变数目增加;地方品种从中部地区向南方和北方扩散过程中则表现为有害突变数目降低趋势(图2)。使用基因组最佳线性无偏预测模型(GBLUP)评估有害突变和随机选择的SNP对抗大豆胞囊线虫2号、3号生理小种和抗大豆花叶病毒表型的影响,发现有害突变的变异解释度增加了4.28%~47.9%(图3)。该研究为通过基因编辑技术清除大豆基因组有害突变以及亲本选择提供了理论和材料支持。 - 【Nature Com】福建农林大学揭示uORF自然变异导致大豆磷效率的多样性
该研究利用前期收集的大豆磷效率应用核心种质资源,开展了全基因组重测序获得了高密度的分子标记;并开展了田间表型鉴定,获取了磷效率相关的表型指标。然后针对植株磷含量进行了全基因组关联分析(GWAS),没有鉴定到显著且背景干净的关联信号;考虑到磷含量与植株总根长的相关性,为了提高GWAS的检测力,研究者引入总根长作为协变量再次对磷含量进行GWAS, 发现在20号染色体上出现了一个新的显著关联位点。研究者推断该位点不影响根长,是通过影响根的吸收效率从而影响磷的吸收量。基于此衍生了一个新的磷吸收效率的表型指标,即单位根长的磷吸收量(植株磷含量/总根长),针对该指标开展GWAS,鉴定到一个显著且背景干净的关联信号,正是上述位于20号染色体的位点,命名为CPU1 (component of phosphorus uptake 1)。 - 南京农业大学揭示通过光合作用提高大豆产量的新机制
JIPB近日在线发表了南京农业大学国家大豆改良中心/作物遗传与种质创新国家重点实验室喻德跃教授课题组题为“GmFtsH25 overexpression increases soybean seed yield by enhancing photosynthesis and photosynthates”的研究论文。该研究首先应用大豆自然群体和重组自交系群体开展光合作用相关性状的遗传解析,在大豆18号染色体上鉴定到一个调控大豆光合作用的遗传位点,分析发现GmFtsH25可能为该位点的候选基因。通过转基因功能验证,发现过表达GmFtsH25能够显著提高大豆植株光合速率和光合产物的积累,进而提高大豆产量,而GmFtsH25敲除突变体的光合速率和光合产物积累明显受到抑制,大豆产量显著降低(图1)。进一步分析发现,GmFtsH25与GmLHCa2之间的相互作用可能有助于优化叶绿体结构,提高光能利用效率(图2)。此外,对1193份大豆种质的基因组序列分析表明,GmFtsH25的优异单倍型(Hap-I)在驯化中被保留,在育种中得到应用(尤其在美洲地区)。综上,此研究揭示了GmFtsH25调控光合作用的分子机制,为通过提高光合作用增加大豆产量提供了重要的理论依据和育种材料。 - 华中农业大学在大豆生物固氮研究中取得新进展
生物固氮为全球植物提供了75%的氮素。ROS过去一直被认为是植物代谢过程中的副产品,然而,越来越多的证据表明,ROS也是细胞信号转导和调控的重要组成部分。豆科植物中的ROS通过直接或间接作用诱导结瘤基因的表达,是根瘤形成以及固氮的重要信号分子。如何调控ROS水平,诱导豆科植物结瘤,从而延长结瘤时间,成为了生物固氮研究的新热点。纳米酶作为纳米技术与生物学之间的桥梁,为生物固氮研究提供了独特的解决方案。 - 黑龙江省农科院在寒地野生大豆花期调控及疫霉根腐病抗性基因研究方面取得进展
黑龙江省农业科学院寒地野生大豆研究团队,以野生大豆为研究材料,以拓宽大豆遗传基础为研究目标,历时三十余年,围绕寒地野生大豆种质资源考察收集保存、原生境和异地繁殖保护、优异性状评价及基因挖掘,桥梁材料创制及育种应用等内容开展了全面深入的研究。近日,团队在野生大豆调控花期和疫霉根腐病抗性基因研究方面取得新进展,研究成果相继在BMC Genomics和Current Issues in Molecular Biology上发表。 - 华南农业大学年海教授课题组通过重组自交系群体挖掘大豆抗炸荚性相关QTL的研究
该研究以在炸荚性状上具有极端表型差异的两个重组自交系群体(CY,华春2号×瓦窑黄豆;GB,桂早1号×巴西13)为材料,对与大豆抗炸荚性状相关QTL进行了鉴定。共检测到14个与大豆炸荚相关的QTL。其中qPS01-1、qPS03-2、qPS05-1、qPS07-1和qPS16-1能在多个环境中被检测到,为环境稳定的QTL,且qPS16-1为本实验所定位到的与大豆炸荚性状相关的主效QTL,通过基因分型最终将qPS16-1的物理区间缩小到了99 Kb(29933890-30033727 bp),目标区间共有8个候选基因,包含一个已被前人鉴定的功能基因Glyma.16g141400,该基因被命名为Pdh1,Pdh1能够通过增加干燥荚果壁的扭曲力促进荚果开裂。结合GO富集分析和转录组测序结果,从另外4个稳定的新QTL位点(qPS01-1、qPS03-2、qPS05-1和qPS07-1)内筛选出9个差异表达基因,其具体的功能还要等待下一步的验证。 - 中国科学院遗传与发育生物学研究所田志喜课题组解析大豆全景遗传重组特征,助力大豆分子设计育种
田志喜课题组联合多家单位利用本课题组开发的高密度SNP芯片对构建的多套大豆重组自交系进行了高通量基因型鉴定,构建了高精度的重组图谱(平均重组区间小于等于2 kb)。研究发现,同源重组事件在染色体上呈现不均匀分布,重组率与基因密度呈正相关关系,与转座子密度、GC含量与结构变异分布呈负相关关系。通过对全基因组重组断点分析发现,其存在一定的保守性,富集于 Poly-A和富含AT的基序,进一步分析发现同源重组更容易发生在表达活跃基因的启动子区域。ATAC-seq实验分析发现,大豆中同源重组事件趋向在开放的染色质区域上产生;CUT&Tag和组蛋白修饰测序分析发现,同源重组断点富集于活跃型组蛋白修饰区域,说明活跃型的组蛋白修饰可能会促进同源重组的形成。此外,通过遗传图谱分析,定位了影响大豆同源重组频率的遗传位点,为解析大豆自然材料间同源重组变异的分子遗传机制奠定了基础。该研究为大豆分子设计育种奠定了重要的理论基础。 - 华南农业大学年海教授与合作者鉴定到大豆抗白粉病基因GmRmd1
近日, Plant Communications在线发表了华南农业大学年海教授、葛良法教授与王应祥教授等题为GmRmd1 encodes a TIR-NBS-BSP protein and confers resistance to powdery mildew in soybean的研究论文,报道他们首次鉴定到大豆抗白粉病基因Rmd1(Resistance-to-M. diffusa 1),填补了大豆抗白粉病育种实践与理论研究间的空白。 - 大豆和拟南芥同源基因对甜菜孢囊线虫的侵染表现相反功能
该研究揭示了2对大豆和拟南芥同源基因(GmSNAP18/ AtSNAP2 和 GmSHMT08/ AtSHMT4)转基因拟南芥对甜菜孢囊线虫表现相反的反应,并证明GmSNAP18和AtSHMT4,及GmSHMT08和AtSNAP2均能互作,另外,这2对转基因拟南芥的AtPR1和AtSHMT4表达模式不同,这些可能和拟南芥对甜菜孢囊线虫的敏感性关联。 - 广州大学发现GIGANTEA同源基因E2家族成员冗余地调控大豆开花与产量
该研究首先通过创制单突和多突变体材料发现,E2家族基因功能冗余却不均衡的调控大豆开花,其中E2起主要作用,两E2 Like基因依赖于E2行使功能。在高纬度地区e2突变体促进开花与产量提高,为大豆育种提供了优异等位变异。分子机制解析发现,E2家族成员之间可以形成同源或者异源二聚体激活豆科特有的转录因子E1家族基因(E1,E1La,E1Lb)转录。遗传关系研究发现E2调控大豆光周期开花与产量完全依赖于E1家族成员。 - 作科所解析双功能锌指蛋白转录因子基因GmZFP7调控大豆异黄酮积累的分子机制
该研究前期利用构建的大豆异黄酮含量显著不同重组自交系群体,结合SLAF-seq重测序技术,构建了大豆高密度连锁图谱(Li et al. 2014, BMC Genomic),结合多年多点的异黄酮表型数据,采用ICM mapping方法,在20号染色体精细定位到一个多年多点可稳定调控异黄酮积累的主效QTL位点qIF20-2,表型解释率20%左右,该连锁区间全长0.53cM,对应的物理图谱长度为243.72 kb(Pei et al. 2018, Crop J),包含28个候选基因,其中17个基因在双亲中存在非同义突变,经转录组表达分析发现有2个转录因子基因在双亲中存在显著差异,一个是MYB类转录因子基因存在一个非同义突变SNP位点,另一个是锌指蛋白类转录因子基因存在2个非同义突变SNP位点,经转基因和基因编辑鉴定,发现该锌指蛋白类转录因子基因GmZFP7与异黄酮积累显著相关。本研究为大豆异黄酮分子育种提供可靠分子证据。 - 中科院东北地理所在Plant Physiolgoy杂志上发表研究论文,在大豆光周期现象分子调控机制研究上取得重要进展
本研究证实E1编码蛋白具有转录抑制活性。基于RNA-seq比较分析发现,有7个GmMDE(Glycine max MADS-box genes downregulated by E1)基因在E1过表达时显著下调,而在CRISPR/Cas9介导的e1敲除突变株中显著上调。这些GmMDE基因表现出相似的组织特异性和表达模式,包括对光周期、E1表达和E1基因型的响应。以GmMDE05和GmMDE06的启动子为代表进行分析表明,E1直接与其启动子结合以增加H3K27me3水平,从而在表观遗传上抑制其表达。组蛋白H3K27 me3翻译后共价修饰是表观遗传调控的重要方式之一,可以维持基因的沉默状态。 - 华南农业大学在国际知名学术期刊(即时影响因子8.48)上发表研究论文,揭示不同测序方法对大豆耐涝根际微生物机制的影响
该项研究利用Illumina Miseq、LoopSeq和PacBio三代全长测序,研究了涝害对中性和酸性土壤大豆根际细菌群落结构的影响。三种测序方法的结果都表明,涝害胁迫显著改变了两种土壤的根际微生物群落结构,并增加了固氮微生物(Geobacter)的丰度。此外,与16S(Illumina Miseq)测序相比,全长(LoopSeq和PacBio)16S测序具有更高的分辨率。核心微生物和网络模块化分析显示,三种测序方法都发现涝害增加了与氮循环相关微生物的相对丰度,而LoopSeq和PacBio测序方法还发现涝害增加了与磷循环相关微生物的相对丰度。核心微生物和网络模块化分析进一步揭示,不同物种的富集可能在维持细菌群落结构和生态功能的稳定性方面起着核心作用。 - 东北农业大学大豆生物学教育部重点实验室在国际知名期刊上发表研究成果
该研究利用六个7S与11S球蛋白亚基组成各异的大豆为原料,采用碱溶酸沉法制备大豆分离蛋白,设置不同处理因素[不同基因型(SPI-1~6)(300 mg/kg/day)及菲诺贝特(30 mg/kg/day)],灌喂随机分成九组的54只高胆固醇模型鼠。28天后,采血测定血脂、采集肝脏及内脏脂肪,观察其病理学变化,测定肝脏总胆固醇(TC)、甘油三脂(TG)含量及胆固醇代谢相关关键基因的mRNA表达量(图2)。结果表明,大豆基因型影响其SPI的降胆固醇功效,不同7S与11S球蛋白亚基组成SPI的降胆固醇功效差异显著,7S-(α′+α)双缺失型“东农豆360”制备的蛋白粉“SPI-2”可以显著提高高胆固醇模型鼠血清的HDL-C水平。不同亚基组成SPI降胆固醇的分子机理各不相同,qRT-PCR结果显示,灌喂28天“东农豆358”制备的保健蛋白粉“SPI-3”可使高胆固醇模型鼠肝脏中的Cyp7a1基因显著上调表达。 - 山西农业大学在大豆花叶病毒病鉴定领域取得突破
本研究根据不同大豆花叶病毒株系CP基因组序列的多态性,设计环介导等温扩增(Loop-mediated Isothermal Amplification, LAMP)SMV-SC7的特异引物组,通过对其引物组筛选、反应条件优化、特异性和灵敏度检测,该方法检测限可低至10−4ng/μL,并同时利用SYBR GreenI、中性红和羟基萘酚蓝实现实验结果可视化,建立了一种特异性强、灵敏度高、操作简单、经济的LAMP可视化检测SMV-SC7的方法。 - 江苏省农科院在大豆异黄酮生物合成领域取得重要进展
与植物提取或化学合成法相比,利用微生物细胞工厂以廉价底物合成天然化合物具有绿色高效、低成本的优势。该团队通过在大肠杆菌底盘细胞中整合来自酵母、苜蓿和葛根等微生物和植物来源的17个酶,并通过代谢途径模块划分和代谢调控对染料木苷合成途径进行优化,构建了以廉价甘油为碳源的染料木苷生物合成细胞工厂。最终,通过补料分批发酵实现了202.7 mg/L的染料木苷产量,这一产量是豆科植物染料木苷产量的273-1140倍,并达到了迄今为止最高的微生物法大豆异黄酮生产水平。 - 中科院遗传发育所揭示大豆分枝数的调控机制
中国科学院遗传与发育生物学研究所植物细胞与染色体工程国家重点实验室田志喜研究组和广州大学孔凡江课题组研究员合作,通过连续两年对2,400多份大豆自然种质资源的分枝数进行表型鉴定,利用全基因组关联分析挖掘到大豆分枝数主效控制基因Dt2。遗传分析发现,Dt2负调控大豆分枝数,CRISPR/Cas9基因敲除株系分枝数明显增多,小区产量明显提高;过表达株系分枝数显著降低,小区产量明显下降。进一步,通过酵母双杂交、双分子荧光互补和免疫共沉淀实验发现,Dt2、GmAgl22和GmSoc1a蛋白可以两两互作。转录组、生物化学和遗传学分析发现,Dt2能够直接结合到GmAp1a和GmAp1d的启动子上,正调控GmAp1a和GmAp1d的转录,同时GmSoc1a和GmAgl22能促进Dt2对GmAp1a和GmAp1d的转录。由此推演出Dt2调控大豆分枝的分子调控模型:Dt2可结合到GmAp1a和GmAp1d的启动子正调控其转录,GmAgl22和GmSoc1a与Dt2的互作可增强了Dt2的转录激活效应。GmAp1a和GmAp1d抑制大豆分枝。 - 抗大豆孢囊线虫新品种 “黑农531”
经大豆高产品种“合丰55”和抗线虫品种“抗线12号”杂交,以其F1代做父本和抗线虫品种“鹏豆158”杂交获得F1代,继而自交获得F2代,然后连续栽培在黑龙江省大豆孢囊线虫发生严重的大豆田里筛选与选育,并经2年7个点12批次的区域试验和1年6个点6批次的生产试验与测产及大豆孢囊线虫侵染表型试验,最终选育成一个高抗线虫、高产、高油大豆新品种“黑农531”(黑审豆20210004),2021年6月正式投放市场。该品种高抗大豆孢囊线虫,其抗性来源于同为Peking型的“鹏豆158” 和“抗线12号”,并且高产、高油.区域试验平均产量为2805.0公斤/公顷,生产试验平均产量达2751.5公斤/公顷,同比地方对照品种“嫩丰18”和“齐农5号”,产量分别提高12.7%和13.6%;种子平均脂肪含量达22.34%。该品种的栽培与推广将有效减轻大豆孢囊线虫的危害,提高大豆的产量与脂肪含量。 - 河南大学发现根瘤菌侵染触发大豆共生根瘤细胞核内复制的机制
本研究以大豆(Glycine max)和根瘤菌USDA110的共生体系为研究对象,研究人员开发了一系列不同的方法来分离根瘤中侵染细胞(ICs)和非侵染细胞(UCs),然后分别测量UCs和ICs中的DNA含量。他们发现并证明在大豆根瘤中UCs和ICs中均包含4C细胞,根瘤菌主要选择侵染4C细胞,随后再触发4C细胞发生核内复制,产生更高倍性水平的细胞。作者还进一步利用流式细胞术分选了根瘤菌侵染后12天(dpi)和20天(dpi)的大豆根瘤中不同倍性的细胞核并进行RNA-Seq,基因表达的分析结果进一步支持了前面的实验结论,表明根瘤菌对4C细胞的侵染是启动核内复制的关键所在。该研究不仅为深入研究根瘤菌和共生固氮领域的诸多问题提供了重要的启示,而且也为研究核内复制在植物发育过程中的作用提供了一个范式。 - 河南农业大学张丹团队揭示GmEIL4调控大豆磷效率的作用机理
该研究定位克隆到一个新的大豆磷效率相关主要数量性状位点基因GmEIL4,其表达受低磷胁迫诱导,并通过调节根长和结构来提升磷的吸收和积累。 - 全基因组重测序揭示了野生大豆的局部适应和分化的特征
本研究收集了来自中国3个主要农业生态区的185份大豆种质资源,并利用全基因组测序数据分析其基因组多样性,研究当地适应的遗传基础。结果表明,大豆具有清晰的地理种群结构和多种环境因素参与了其遗传分化。种群历史分析显示,三个生态区的大豆在大约100000年前出现分歧,随后其有效种群规模经历了不同程度的扩张。全基因组环境关联鉴定了多个参与大豆局部适应的基因,特别是开花时间和温度相关基因。最后,本研究阐述了大豆局部适应的遗传基础,为大豆的分化提供了新的见解,有助于在目前主要大豆种植区域以外的更广泛地区育种适应气候的大豆品种。 - 中国农科院植保所研发出基于HIGS的双抗大豆新种质材料
该文从克隆到的大豆孢囊线虫几丁质合成酶基因( SCN-CHS )选取催化结构域的一段420bp序列构建了 SCN-CHS 的RNAi质粒,经农杆菌转化获得了3株纯合HIGS转 SCN-CHS 基因大豆新种质材料,T6代HIGS转基因株系(IPP48-7-5,IPP55-8-24和 IPP57-9-2)对大豆孢囊线虫4号生理小种具有高度抗性,显著抑制了线虫的发育,HIGS转基因植株能稳定靶标与沉默 SCN-CHS ,提高对大豆孢囊线虫的抗性。同时,HIGS转基因植株对尖孢镰刀菌( Fusarium oxysporum )引起大豆的镰刀菌枯萎病(Fusarium wilt disease)具有中度抗性。 - 中国农业科学院解析双功能锌指蛋白类转录因子GmZFP7调控大豆异黄酮积累的分子机制
该研究发现了一个锌指蛋白类转录因子基因GmZFP7。实验表明,过表达该基因可使发状根中的异黄酮比对照提高1.15-3.79倍,而沉默该基因可使异黄酮含量降低到27%。分析发现,该基因具有可以促进异黄酮合成过程中关键酶GmIFS2基因表达,同时抑制黄烷酮合成路径中的GmF3H1基因表达的双功能。为了进一步验证该基因功能,研究人员获得了基因编辑敲除和过表达转化植株,在敲除该基因植株的叶片和种子中,该基因的表达水平和异黄酮含量显著降低,同时GmF3H1的表达量提高;而在过表达植株叶片和种子中结果反之。代谢组分析表明,在基因敲除种子中可检测到57种酚类代谢产物,其中24种与对照存在差异,特别是在异黄酮合成路径中的上游产物含量显著升高,如苯丙烷路径中的肉桂酸、七叶树素、异甘草酸和根皮苷,黄酮类路径中的大波斯菊甙、毛地黄黄酮、芹黄素,以及黄烷酮路径中的柚皮苷和柚皮素;而黄烷醇合成路径中的槲皮素和烟花甙含量显著降低,芦丁、异鼠李亭甙和野樱素含量显著提高。该研究还基于1557份大豆种质的单倍型分析,鉴定出两个异黄酮含量显著较高的优异单倍型。该研究为大豆异黄酮的分子育种提供了新思路和优异基因。 - 南京农业大学揭示通过光合作用提高大豆产量的新机制
该研究首先应用大豆自然群体和重组自交系群体开展光合作用相关性状的遗传解析,在大豆18号染色体上鉴定到一个调控大豆光合作用的遗传位点,分析发现GmFtsH25可能为该位点的候选基因。通过转基因功能验证,发现过表达GmFtsH25能够显著提高大豆植株光合速率和光合产物的积累,进而提高大豆产量,而GmFtsH25敲除突变体的光合速率和光合产物积累明显受到抑制,大豆产量显著降低(图1)。进一步分析发现,GmFtsH25与GmLHCa2之间的相互作用可能有助于优化叶绿体结构,提高光能利用效率(图2)。此外,对1193份大豆种质的基因组序列分析表明,GmFtsH25的优异单倍型(Hap-I)在驯化中被保留,在育种中得到应用(尤其在美洲地区)。综上,此研究揭示了GmFtsH25调控光合作用的分子机制,为通过提高光合作用增加大豆产量提供了重要的理论依据和育种材料。 - 王学路教授团队在大豆共生固氮领域取得重大突破
这项突破性进展,揭示了大豆根瘤中的新型能量感受器GmNAS1/GmNAP1通过调控根瘤碳源的重新分配,进而调整根瘤固氮能力的分子机制,并表明动物细胞和植物细胞采用各具特色的分子机制感受能量。该机制使豆科植物可以在生长环境改变时,依据其体内碳源的可用性及时调整根瘤固氮效能,从而维持植株体内的碳氮平衡,适应周围环境的变化。这项突破性成果,为发掘自主产生碳源的植物中更多的能量感受器并建立其信号通路提供了范例,将极大促进对细胞和个体水平碳源分配和代谢调控的进化和分子机制的解析,并为未来通过合成生物学方法,设计高效利用作物自身或者周围环境中的碳源,提高共生固氮能力提供了重要理论支撑,为高效固氮作物的分子设计提供了新的思路。 - 刘宝辉课题组综述大豆驯化基因研究进展
总结了调控大豆驯化性状(包括开花、炸荚、休眠、种子硬度、油分和泥膜等性状)相关基因的研究进展。 - 大豆GolS基因家族鉴定及盐旱胁迫下的表达分析
肌醇半乳糖苷合成酶(galactinol synthase, GolS) 是棉子糖家族寡糖(raffinose family oligosaccharides, RFOs)生物合成途径中的关键酶,在植物对非生物胁迫的反应中发挥重要作用。然而,关于大豆(Glycine max)GolS基因家族成员的分子结构特征还未见研究报道。本研究在全基因组水平上鉴定了6个大豆GolS基因家族成员,并对其理化性质、染色体定位、进化关系、基因结构、保守基序、二级结构、三级结构、组织特异性表达模式以及盐和干旱胁迫下的表达量进行了分析。结果表明:6个大豆GolS基因不均匀地分布在4条染色体上,6个大豆GolS蛋白的等电点为5.45−6.08,分子量变化范围为37 567.07−38 817.59 Da,氨基酸数量为324−339 aa;亚细胞定位预测结果发现4个蛋白定位在叶绿体上,2个蛋白定位在细胞质。系统进化树分析表明,大豆GolS基因家族成员在进化树中呈现出两两紧邻的现象,在进化上较为保守。6个基因成员含有的外显子数目为3或4。二级结构和三级结构预测表明,该家族所有成员蛋白质的空间结构主要由α螺旋和无规则卷曲结构组成,有较少的β转角结构和延伸链结构。组织特异性表达分析表明,6个GmGolS家族成员在种子、根、根毛、花、茎、豆荚、根瘤和叶中均有不同程度表达。基于qRT-PCR的表达分析显示,盐旱处理后所有GmGolS基因成员表现出不同程度的上调表达,表明这些基因可能与植物的耐盐抗旱响应有关。本研究结果为后续开展大豆GolS基因的功能解析奠定了基础。 - 中国农科院作科所与长江大学合作解析大豆百粒重与荚型遗传基础并构建基因组选择模型
该研究以栽培大豆中豆41 × 野生大豆(ZYD02878)的重组近交系(RIL)群体为研究材料,基于多环境表型鉴定及遗传连锁图谱,利用多种遗传分析方法挖掘百粒重、荚长和荚宽相关QTL,并在此基础上构建了基因组选择(GS)模型。
2021
- 江苏省农科院揭示了GmAOC4基因通过促进更多 JA 积累调节大豆种子萌发的分子机制
揭示了GmAOC4基因通过促进更多 JA 积累调节大豆种子萌发的分子机制,该研究对提升大豆种源活力具有重要应用价值。 - 广州大学在Cell子刊发表研究论文,在大豆适应性的遗传机制研究取得重要进展
该研究通过图位克隆和群体遗传学分析发现,大豆两个 FT 同源基因,FT2a 和 FT5a,分别编码两个 LJ 性状 QTL 位点,进一步研究发现,FT2a和FT5a的变异能抑制 AP1 的转录,从而延迟短日照条件下大豆的开花和成熟,研究还发现,ft2a 和 ft5a 的单突变体表现出严重的遗传补偿反应 (Genetic compensation response),开花期延迟相对较少,而 ft2a ft5a 双突变则可以打破这种补偿反应,表现出增强的 LJ 表型,并在低纬度地区的短日照条件下转化为更高的产量。 - 南京农业大学在大豆质核互作雄性不育机理研究中取得重要进展
该研究对不育系NJCMS1A的育性恢复基因进行遗传定位,确定育性恢复基因位于从Glyma.16G161600到Glyma.16G163400这19个基因的基因组区段。在含有育性恢复基因的恢复系NJCMS1C和不含育性恢复基因的不育系NJCMS1A及其保持系NJCMS1B中分别扩增这19个基因的基因组序列,通过序列比对初步缩小候选育性恢复基因的范围,确定育性恢复基因可能是PPR家族基因GmPPR576(Glyma.16G161900)和GmPPR552(Glyma.16G162100)或二者之一。在多个恢复系、不育系和保持系中获得候选育性恢复基因的序列,通过序列比对、单倍型分析和系统发育分析进一步确定候选育性恢复基因可能是GmPPR576(Glyma.16G161900)。大豆N8855是NJCMS1A雄性不育细胞质供体,N8855的育性正常,说明其本身携带育性恢复基因,具有天然的CMS/Rf系统,如果破坏其中的育性恢复基因Rf的功能,则可导致N8855发生雄性不育。利用CRISPR/Cas9技术成功敲除大豆N8855中的候选育性恢复基因GmPPR576,基因编辑植株发生雄性不育,证明候选育性恢复基因GmPPR576是NJCMS1A的育性恢复基因。育性恢复基因的确定对于通过“三系”杂交种生产体系实现大豆杂种优势利用具有重要的理论和实践意义。 - 中国农业科学院作科所在大豆耐干旱和盐胁迫研究中取得重要进展
研究人员在大豆干旱RNA-seq数据中发现NF-Y家族转录因子基因在干旱胁迫下的表达发生了显著变化,其中多个NF-YC亚基基因的表达水平上调显著。表达模式分析进一步发现,大豆NF-YC亚基家族成员GmNF-YC14在干旱、盐胁迫以及ABA处理下能够同时被显著诱导上调表达。因此GmNF-YC14基因被选出进行深入研究。研究人员利用农杆菌介导的稳定遗传转化技术,将GmNF-YC14导入大豆基因组中形成过表达植株。同时为了进一步分析GmNF-YC14基因在大豆响应非生物胁迫中的作用,研究人员构建了CRISPR/Cas9介导的GmNF-YC14基因编辑大豆突变体材料,并筛选到两个不同位点突变的突变体材料,其中,Gmnf-yc14-KO1突变体在PAM位点前3个碱基处丢失了2个AC碱基,Gmnf-yc14-KO2突变体在PAM位点前4个碱基处丢失5个碱基CGATA,且最终导致氨基酸序列发生改变(图1)。 - 普渡大学马渐新组等揭示大豆疫霉菌广谱抗性基因
通过基于无间隙序列的精细定位和表达分析,将一个 27.7 kb 的 NLR 基因确定为Rps11的候选基因。为了进一步证明Rps11的功能,该研究通过开发Rps11转基因品系并检查它们对病原体的反应。研究将 PAtUbi 3::CDS- Rps11载体体引入优良大豆品种 93Y21 中。研究表明,带有转基因的 T 2后代显示出对这三个种族的抗性,其抗性水平与源自作图群体的纯合 F 5 RIL ( Rps11/Rps11 )显示的水平相似。 - 盖钧镒院士团队鉴定到对多个大豆花叶病毒株系具有广谱抗性的转录因子GmCAL
该研究从抗SMV大豆品种科丰1号中克隆了MADS-box转录因子GmCAL的全长编码序列。通过对该基因的SMV诱导表达分析、组织表达分析和亚细胞定位,并利用VIGS沉默和过表达体系鉴定了该基因功能。 - 南京农业大学王源超团队利用CRISPR/LbCpf1系统在大豆上实现高效多基因编辑及染色体大片段删除
该研究在大豆中建立了一种高效、便捷的CRISPR/LbCpf1多基因编辑系统,实现了8个靶标的同时编辑,并在大豆中成功实现了染色体小片段(<1kb)、大片段(~10Kb)和超大片段(>10Kb)的删除。 - 历时10年!我国学者发表Cell, Nature, NG, NBT等论文,在大豆基因组方面取得一系列重要成果!
研究者利用全基因组鸟枪法对大豆进行全基因组测序,利用大豆栽培品种Williams 82品种大豆家系的444个重组自交系构建遗传图谱用来辅助组装,最终组装后的基因组大小为994Mb,ContigN50为189.4 Kb,ScaffoldN50达47.8 Mb,其中有397条Scaffold组装并锚定到20条染色体水平,组装基因组中确定了4991个SNP和874个SSR,并预测出46430个蛋白编码基因,重复序列占到整个基因组的59%。 - 中科院遗传发育所在Plant Biotechnology Journal上发表研究论文,发现大豆耐盐新机制
这项研究揭示了大豆GmNFYA通过调控组蛋白乙酰化提高耐盐性的机制,对于大豆耐逆育种具有重要借鉴和潜在应用价值。 - 广州大学在Cell子刊发表研究论文,在大豆适应性的遗传机制研究取得重要进展
该研究通过图位克隆和群体遗传学分析发现,大豆两个 FT 同源基因,FT2a 和 FT5a,分别编码两个 LJ 性状 QTL 位点,进一步研究发现,FT2a和FT5a的变异能抑制 AP1 的转录,从而延迟短日照条件下大豆的开花和成熟,研究还发现,ft2a 和 ft5a 的单突变体表现出严重的遗传补偿反应 (Genetic compensation response),开花期延迟相对较少,而 ft2a ft5a 双突变则可以打破这种补偿反应,表现出增强的 LJ 表型,并在低纬度地区的短日照条件下转化为更高的产量。 - 干旱胁迫下不同品种大豆籽粒发育期蛋白质含量积累的研究
耐旱品种晋豆21旱处理后,农艺性状变化不大,含水量变化基本相对应,籽粒蛋白质积累规律仍与正常情况基本一致,说明其具有较强的抗旱性,种子内部也具备相对完善的抗旱机制,可作为抗旱育种的优质种质资源。大豆抗旱性属于复杂的生物性状,受多个基因精细严谨调控。晋豆21的强抗旱性已得到认可,但其因种子细小,推广种植受到制约,应从基因表达水平上进一步研究其抗旱机制,为后期探究、筛选、创制新的优质抗旱品种提供基础。 - 福建农林大学在大豆根瘤发育调控机制研究中取得进展
该研究建立了大豆根瘤发育的独特的细胞生物学体系,可视化的呈现了大豆根瘤的发生和发育过程不同阶段生长素的分布和浓度梯度的建立。
2020
- PBJ | 又一篇大豆基因编辑论文!中国农科院作科所成功实现大豆基因单碱基替换,创造晚花表型突变体!
该研究通过组合Cas9n(D10A)切口酶、大鼠胞苷脱氨酶(APOBEC1)和尿嘧啶糖基化酶抑制剂(UGI),建立了大豆单碱基编辑技术体系,并成功实现了大豆开花调控关键基因GmFT2a和GmFT4的单碱基定向替换,效率分别为18.2%(4/22)和6.0%(2/34)。在GmFT2a靶位点实现了C→T和C→G两种类型的单碱基替换、在GmFT4靶位点实现了C→G单碱基替换。单碱基替换突变可稳定遗传,筛选获得的GmFT2a单碱基替换纯合突变系,在长日照(16小时光照/8小时黑暗)和短日照(12小时光照/12小时黑暗)条件下均表现出晚花表型,为大豆重要农艺性状的定向改良提供了新技术、新路径。 - 高水平论文 | 作科所孙君明研究员团队解析我国1025份大豆种质脂肪酸组分的区域分布规律
该研究收集了我国三个不同大豆生态区涵盖全国29个省份的1025份大豆种质,分别于2017-2018年种植在北京、安徽和海南三个不同地点,采用气相色谱技术分析多年多点的大豆种质的脂肪酸组成,探究不同来源大豆种质的脂肪酸区域分布特点,以期为大豆营养品质育种提供理论指导,同时也为大豆加工业提供可靠的原料信息。 - 【MP/PBJ】两篇论文同时揭示生物钟基因在大豆驯化中的作用机制
通过不同策略克隆了与生物钟调控和大豆驯化相关的基因 GmPRR3b/GmPRR37 , 从多个侧面阐明了该基因调控大豆生物钟和生育期的功能机制,并揭示其自然等位变异与早花和高产性状的紧密联系及在大豆驯化和品种改良中的广泛利用。该研究不仅对理解大豆驯化过程具有重要的科学意义,同时为选育应对气候变化和适应高纬地区种植的大豆品种提供了理论依据和重要靶点。 - 蛋白质沉淀法去除干扰大豆种子中蛋白结合色氨酸定量的碳水化合物
Tryptophan is one of the nine essential amino acids in humans that can only be obtained through diets and supplements. It is a precursor to many biological processes, such as serotonin, melatonin, kynurenin, and niacin (nicotinamide) vitamin synthesis. The content of tryptophan in foods, such as soybean is an important indicator of nutritional value. Therefore, accurate quantification of tryptophan in soybean is crucial to soybean nutritional improvement. Quantification of soybean protein-bound amino acids first involves acid hydrolysis of total protein to liberate amino acids. However, tryptophan quantification following acid hydrolysis is difficult or impossible due to its reactions with soybean carbohydrates. Therefore, removal of carbohydrates from soy proteins prior to acid hydrolysis is necessary. In this study, we compared four common protein precipitation methods (i.e., methanol, acetonitrile, acetone, and trichloroacetic acid (TCA) protein precipitation methods) to determine the best method to separate soy proteins from carbohydrates, and concluded that acetone provided the highest recovery of soy proteins. Tryptophan content in the precipitated proteins was determined after acid hydrolysis of the proteins using liquid chromatography–tandem mass spectrometry multiple reaction monitoring (LC–MS/MS–MRM). No significant difference in the tryptophan content was found among proteins precipitated with methanol, acetonitrile, and TCA, suggesting that these precipitated proteins have similar compositions. A slightly lower, but statistically significant tryptophan content was found in the acetonitrile-precipitated proteins, suggesting that these proteins contain slightly higher glycosylated proteins.
2018
- 华中农业大学发现GmmiR156b调控大豆理想株型和高产
该研究首次明确了GmmiR156b是调控大豆理想株型和高产的主效基因,揭示了调控植物分枝形成和发育的miR156b-SPL-WUS的新机制, 也为培育高产大豆提供了具有重要育种价值的基因资源和理论依据
2017
- 大豆百粒重调控基因首次被中国科学家克隆
首次克隆了大豆百粒重QTL基因,揭示了基因功能,鉴定了优异等位变异。通过分析衍生于野生大豆ZYD7和栽培大豆HN44的1036份重组自交系材料,鉴定了一个种子百粒重增加的株系R245。随后对198份自交系材料和2个亲本进行了全基因组重测序,获得了高质量的SNPs。以上述SNPs作为分子标记,构建了大豆遗传图谱并利用多年多点的数据定位了调控种子百粒重和油分含量的QTL位点。其中调控种子百粒重的有14个位点而控制油分含量的有3个位点