Tutorial of LIRBase
LIRBase is a database with comprehensive collection of long inverted repeats in 427 eukaryotic genomes.
Using IRF (https://tandem.bu.edu/irf/irf.download.html), we identified a total of 6,789,791 long inverted repeats in the whole genomes of 427 eukaryotes, including 297,317 LIRs in 77 invertebrate metazoa genomes, 1,902,296 LIRs in 142 plant genomes and 4,585,178 LIRs in 208 vertebrate genomes. LIRBase is deployed at https://venyao.xyz/lirbase/ for online use.
The homepage of LIRBase displays the main functionalities of LIRBase (Figure 1).
- Browse long inverted repeats (LIR) identified in 427 eukaryotic genomes for the sequences, structures of LIRs, and the overlaps between LIRs and genes.
- Search LIRBase for long inverted repeats in a specific genome by genomic locations.
- Search LIRBase for long inverted repeats in a specific genome by the identifiers of long inverted repeats.
- Search LIRBase by sequence similarity using BLAST.
- Detect and annotate long inverted repeats in user-uploaded DNA sequences.
- Align small RNA sequencing data to long inverted repeats of a specific genome to detect the origination of small RNAs from long inverted repeats and quantify the expression level of long inverted repeats.
- Perform differential expression analysis of long inverted repeats or small RNAs between different biological samples/tissues.
- Identify protein-coding genes targeted by the small RNAs derived from a LIR through detecting the complementary matches between small RNAs and the cDNA sequence of protein-coding genes.
- Predict and visualize the secondary structure of potential hpRNA encoded by a LIR using RNAfold.
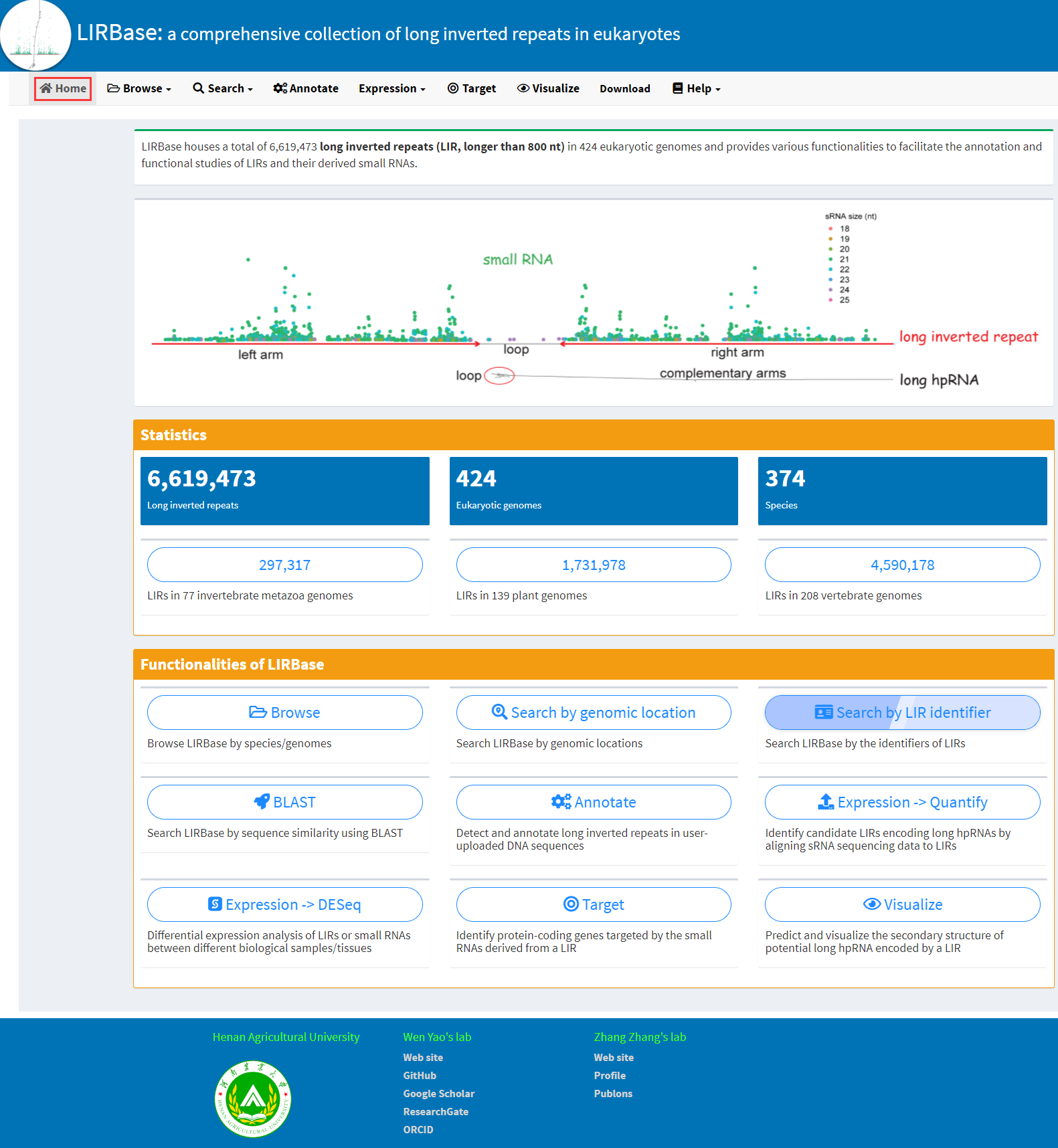
Figure 1. The homepage of LIRBase.
1. Browse LIRBase for long inverted repeats identified in 427 eukaryotic genomes
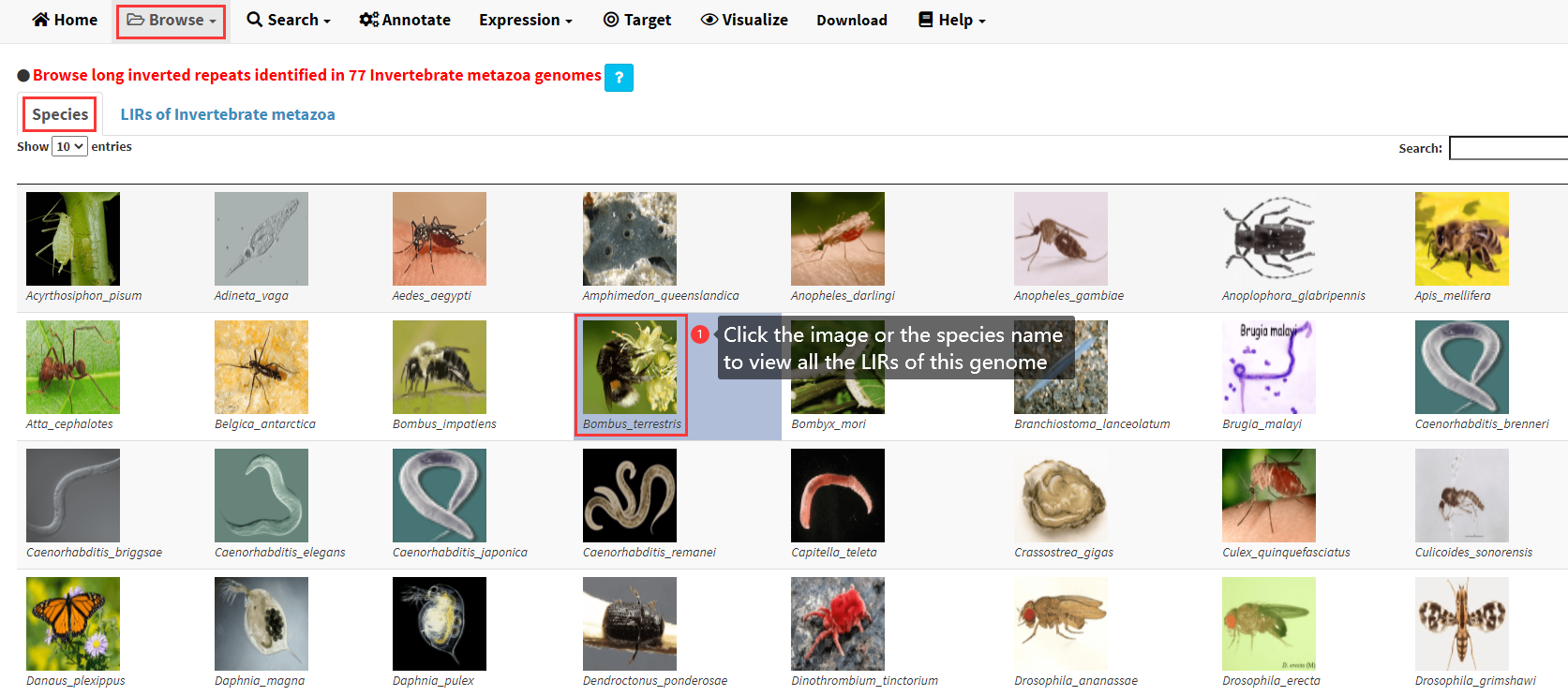
The images and the species names of 77 invertebrate metazoa genomes are listed in the Species panel of the Invertebrate metazoa submenu under the Browse menu (Figure 2). The images and the species names of 142 plant genomes are listed in the Species panel of the Plant submenu under the Browse menu. The images and the species names of 208 vertebrate genomes are listed in the Species of under the Vertebrate submenu under the Browse menu.
Figure 2. Species name and images of 77 Invertebrate metazoa genomes listed in the Species panel of the Invertebrate metazoa submenu under the Browse menu.
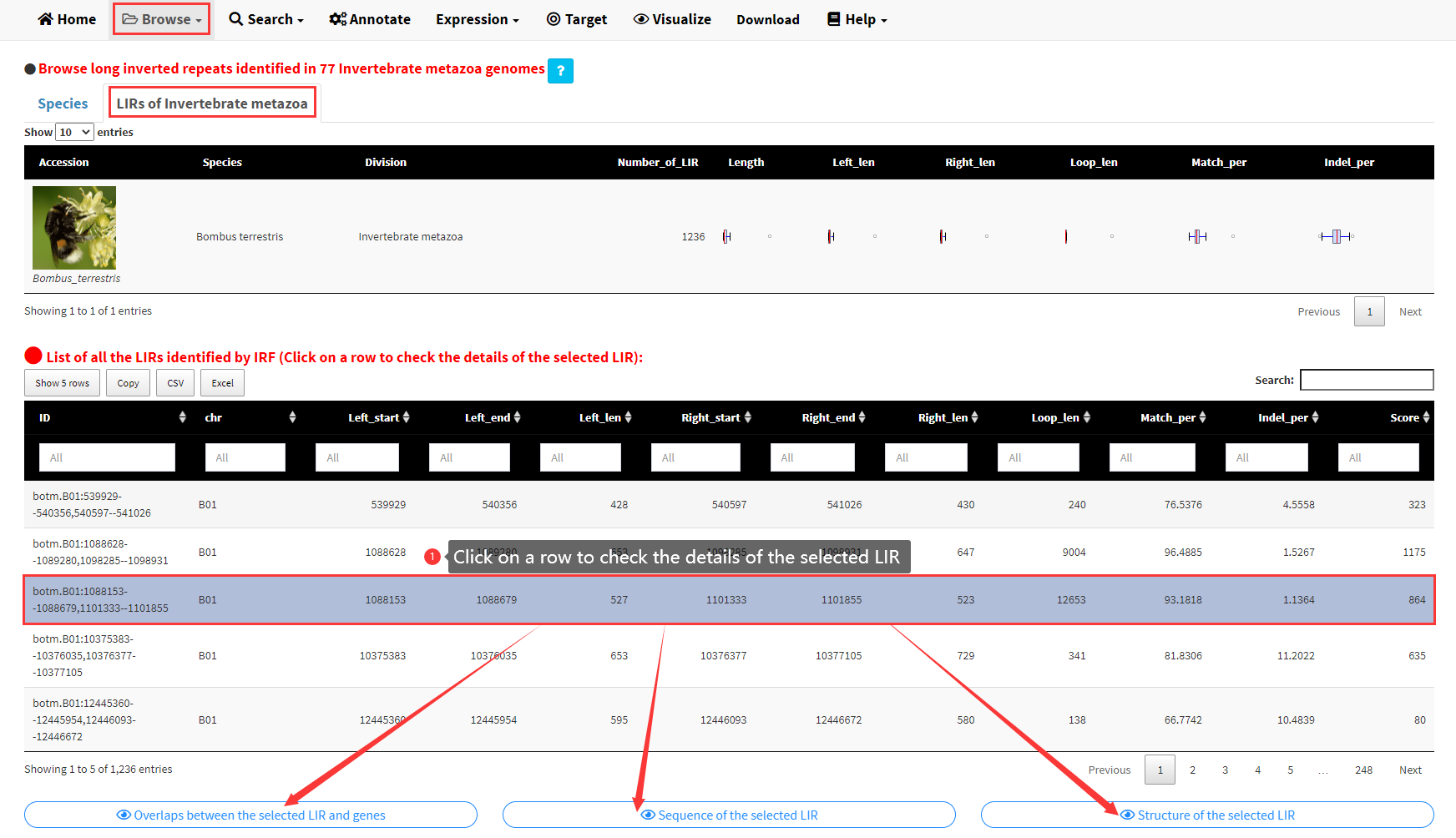
Click of the image or the species name of any genome would take you to the LIRs of Invertebrate metazoa panel of the Invertebrate metazoa submenu under the Browse menu, which displays all the LIRs identified in the selected genome (Figure 3). A brief summary of all the LIRs identified in the selected genome and a table of all the LIRs showing the structure of each LIR are demonstrated in the LIRs of Invertebrate metazoa panel. Three buttons would be displayed below the table if any row of the table was clicked (Figure 3).
Figure 3. List of all the LIRs identified for a selected genome.
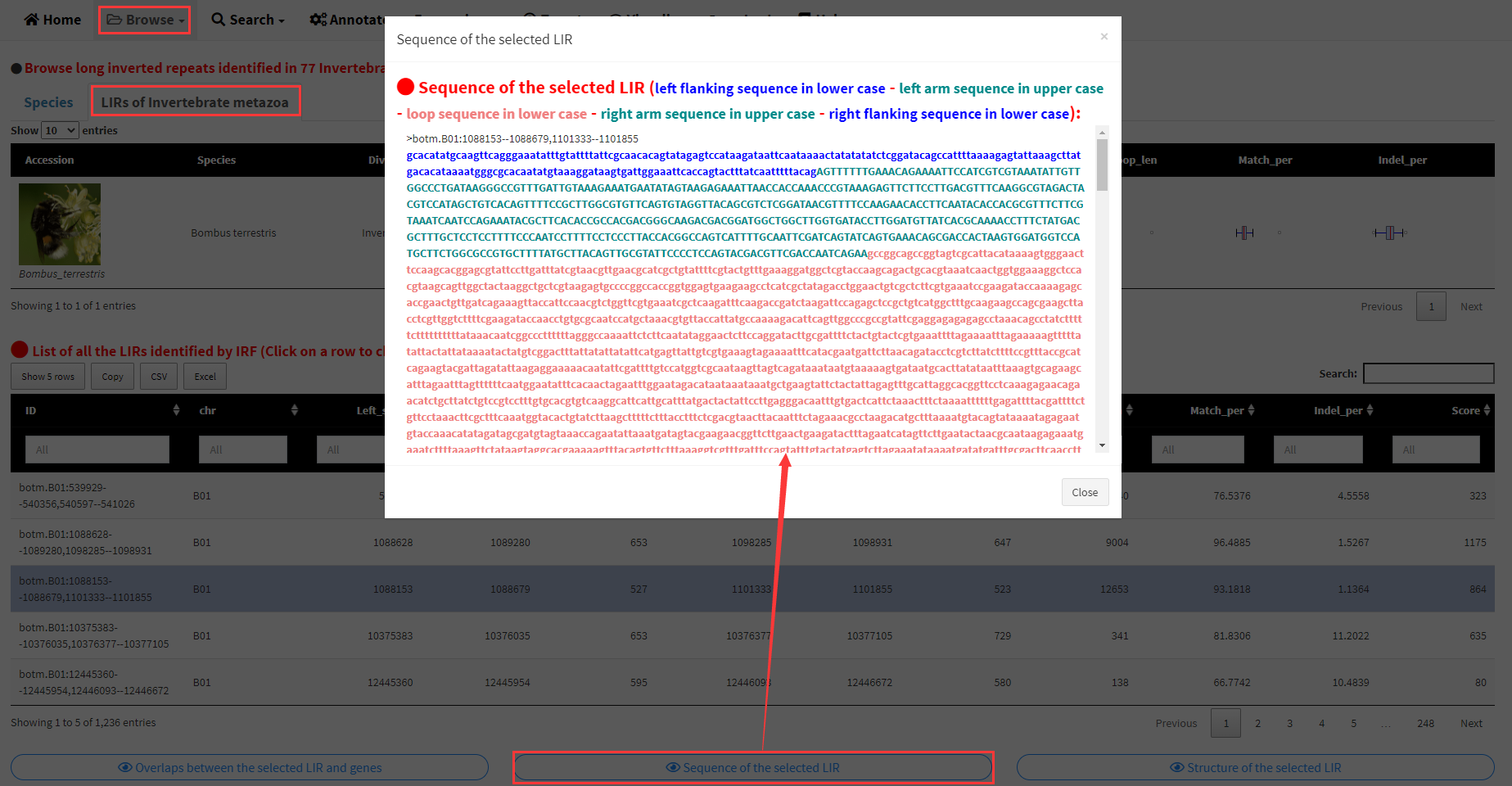
The three buttons below the table can be clicked to display the sequence, structure of the selected LIR and the overlaps between the selected LIR and genes, respectively (Figure 4).
Figure 4. Detailed information of a selected LIR.
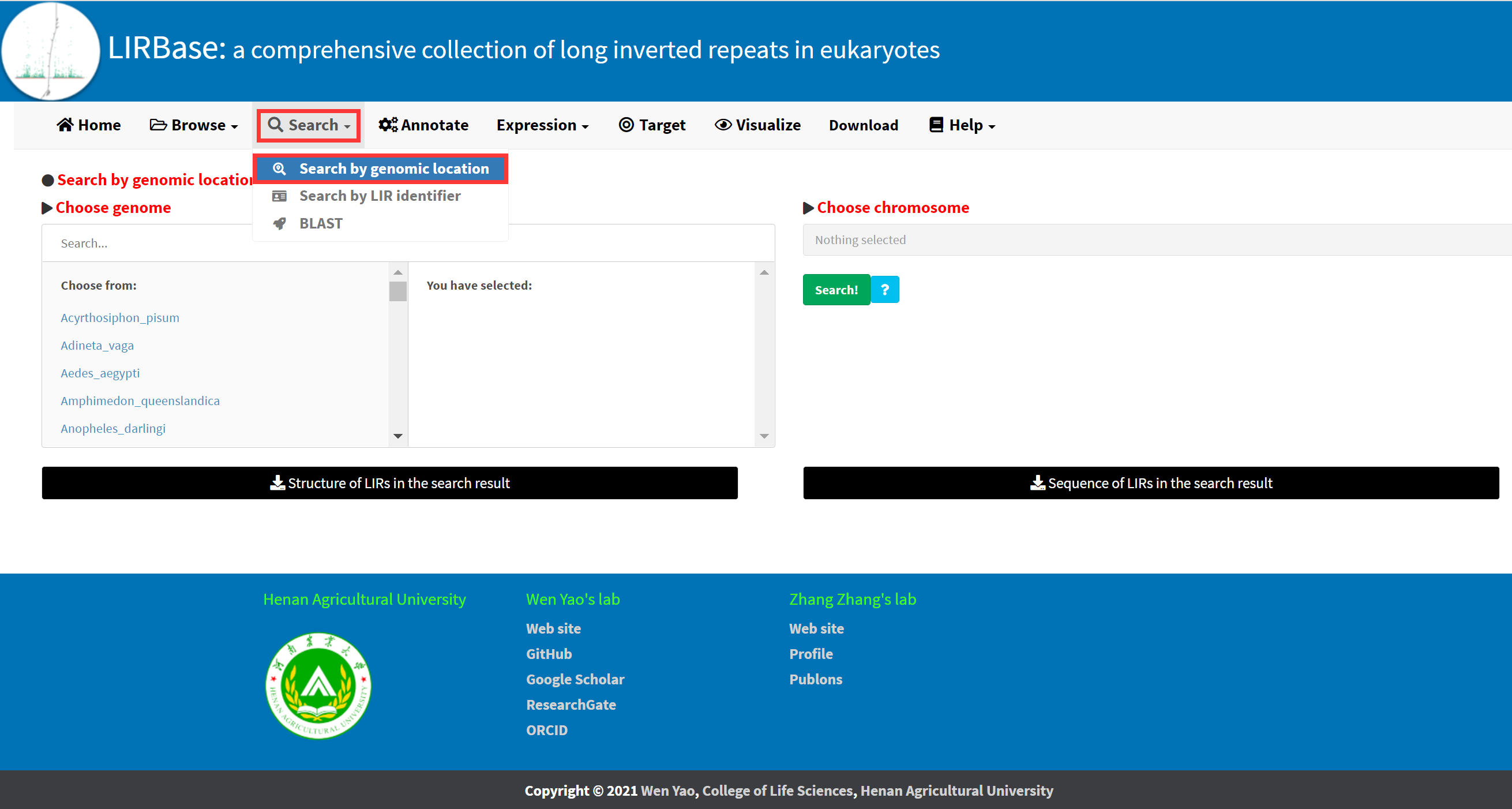
2. Search LIRBase by genomic locations
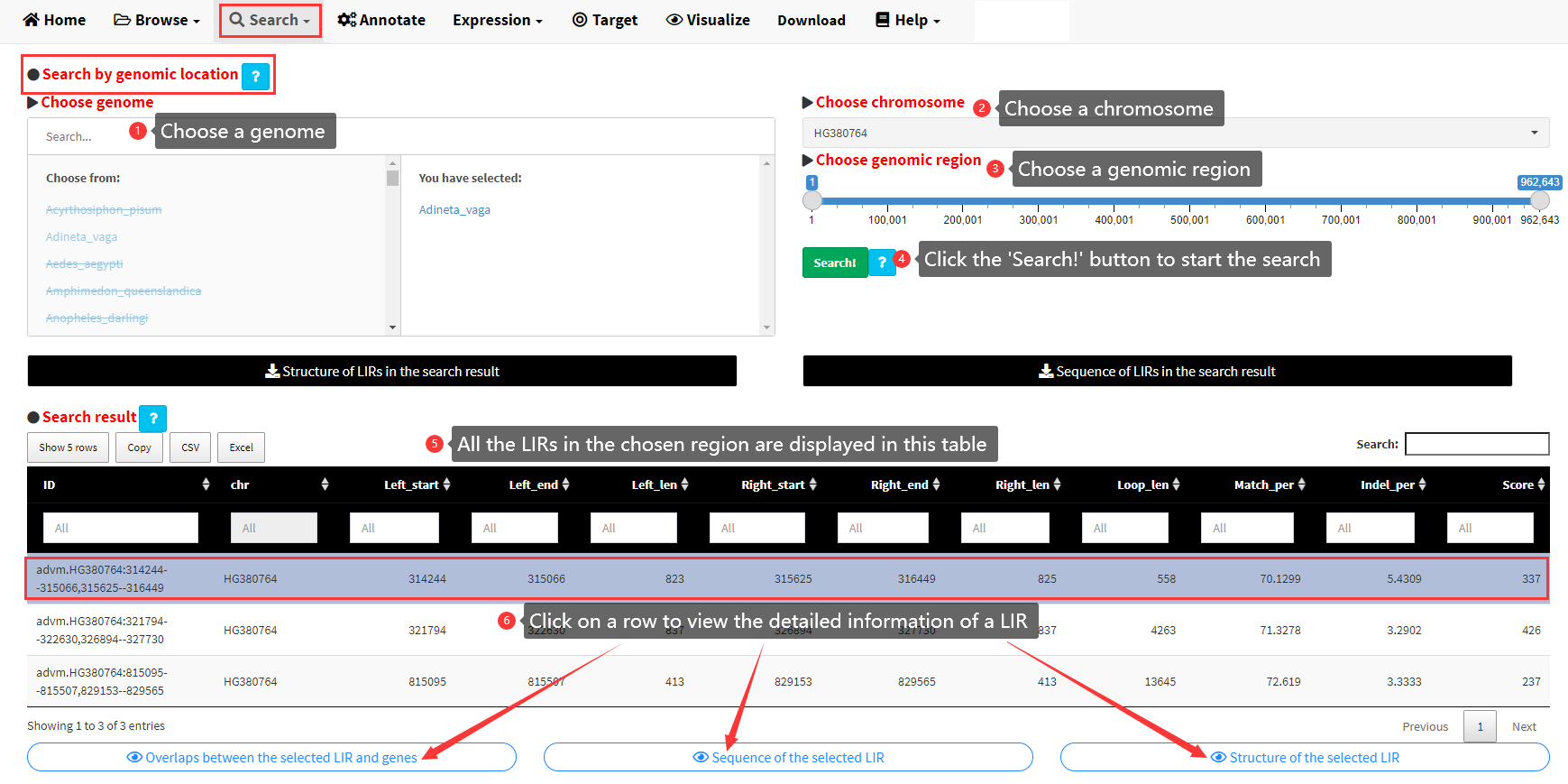
LIRBase allows searching for LIRs identified in any of the 427 eukaryotic genomes by genomic locations (Figure 5).
Figure 5. The Search by genomic location submenu under the Search menu.
The detailed steps to search LIRBase by genomic locations are shown in Figure 6. The search results are displayed as a data table (Figure 6). Each row of the data table represents a LIR in the search result. Three buttons would be displayed below the table if any row of the table was selected. The detailed information of a LIR including the sequence, structure of the selected LIR and the overlaps between the selected LIR and genes, can be viewed by clicking the three buttons (Figure 6)
Figure 6. Steps to search LIRBase by genomic location.
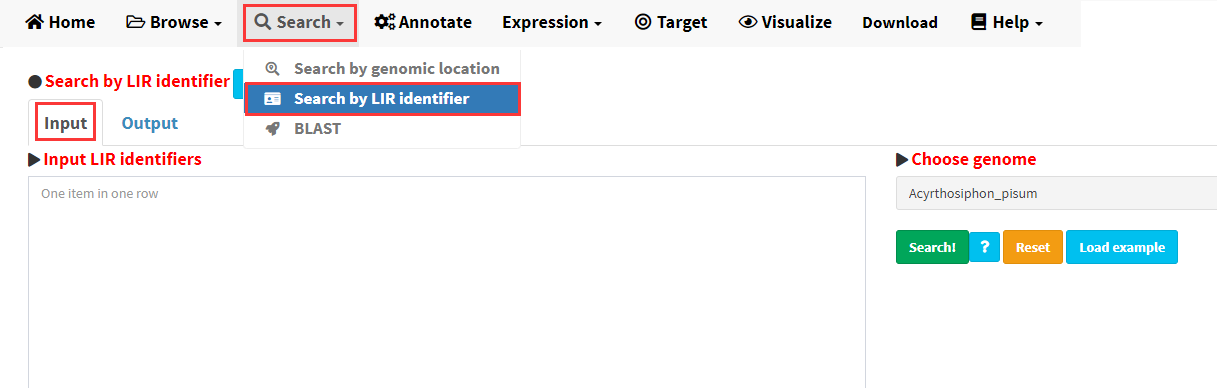
3. Search LIRBase by the identifiers of LIRs
LIRBase allows searching for LIRs identified in any of the 427 eukaryotic genomes by the identifiers (IDs) of long inverted repeats (Figure 7).
Figure 7. The Search by LIR identifier submenu under the Search menu.
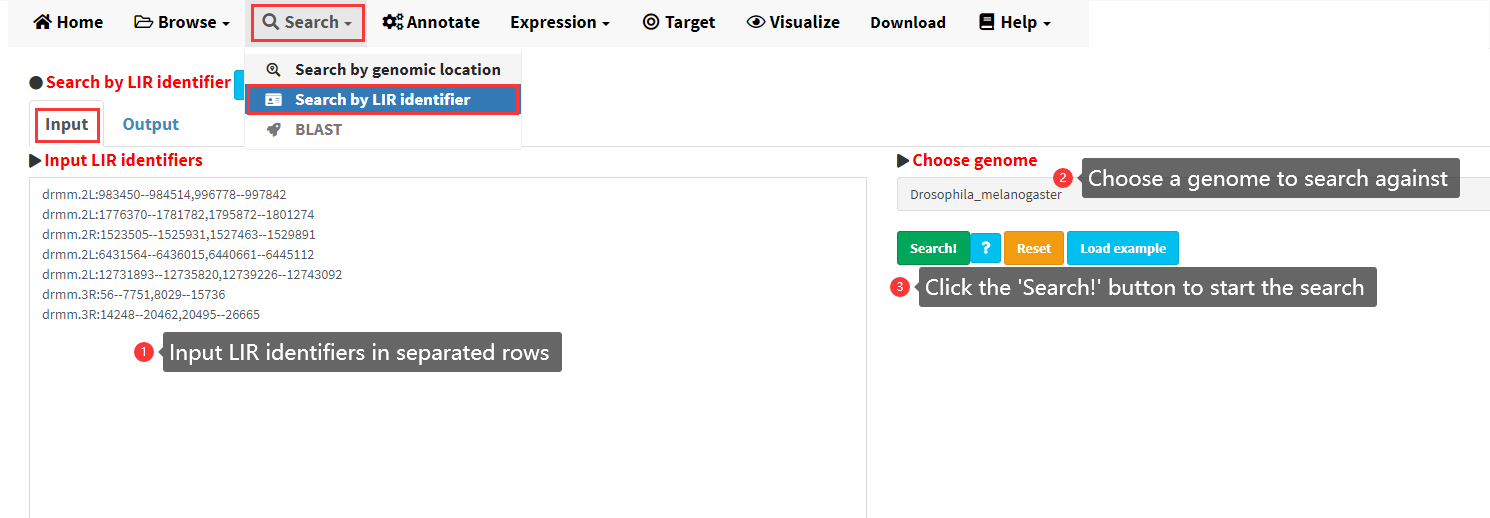
The detailed steps to search LIRBase by the identifiers of LIRs are shown in Figure 8.
Figure 8. Steps to search LIRBase by LIR identifiers.
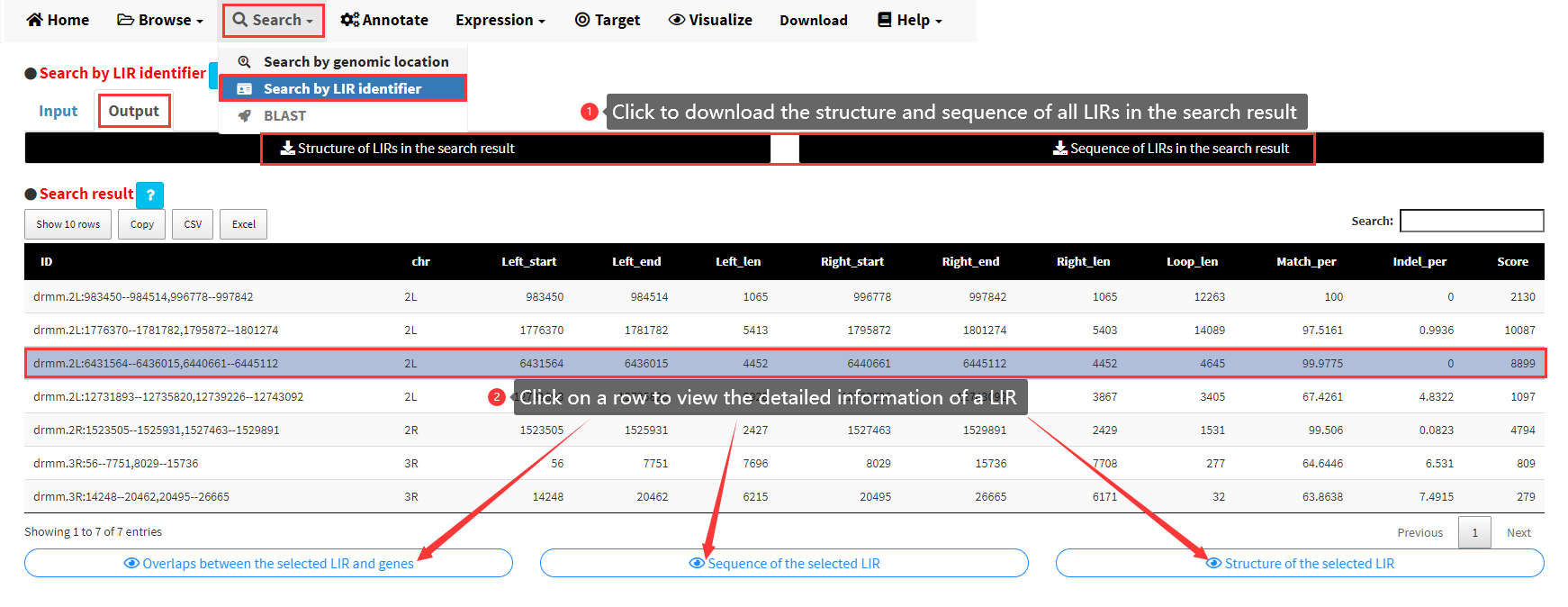
After clicking the Search button in the Input panel shown in Figure 8, the results would be displayed as a data table in the Output panel (Figure 9). The result can also be downloaded by clicking the download buttons on top of the data table. Each row of the data table represents a LIR in the search result. Three buttons would be displayed below the table if any row of the table was clicked. The detailed information of a LIR including the sequence, structure of the selected LIR and the overlaps between the selected LIR and genes, can be viewed by clicking the three buttons (Figure 9).
Figure 9. The Output panel of the Search by LIR identifier submenu.
4. Search LIRBase using BLAST
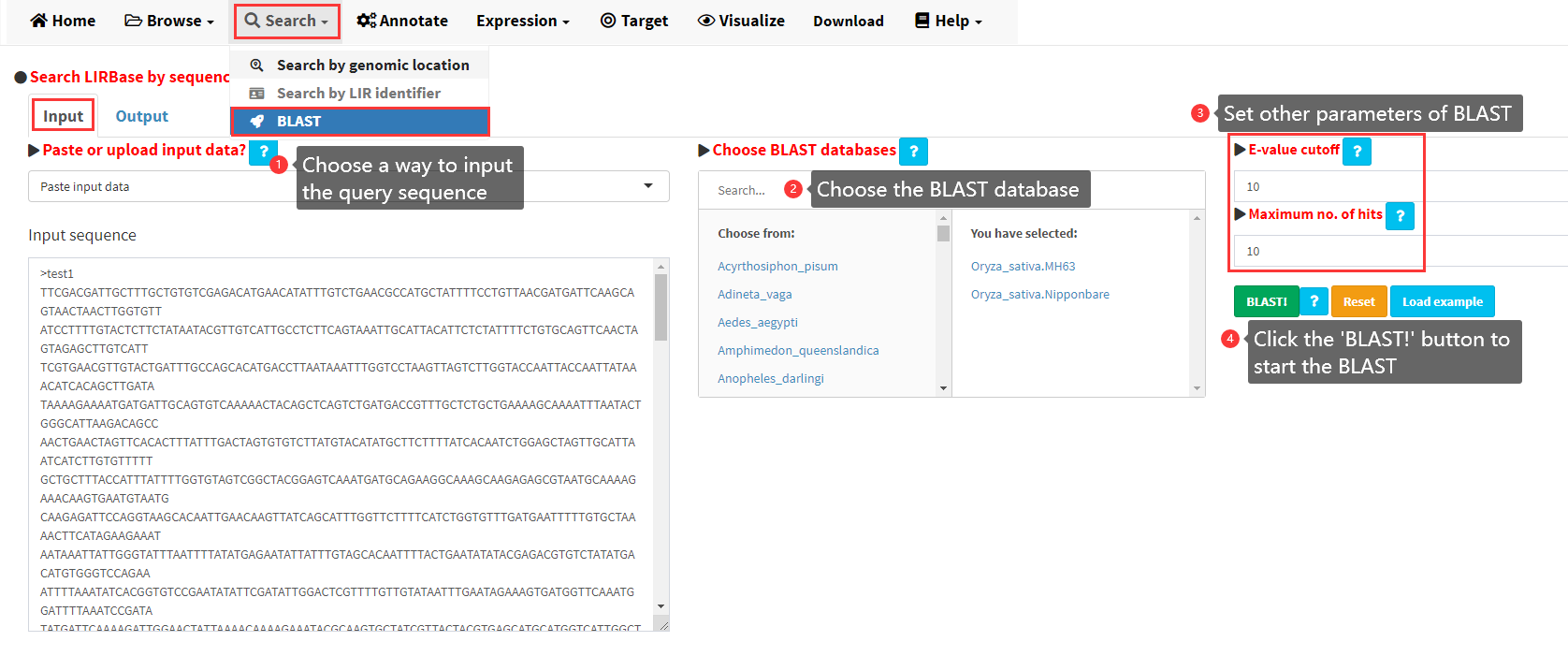
Users can also search LIRBase by sequence similarity using BLAST (Figure 10). A graphical interface was implemented in LIRBase for users to perform BLAST alignment through the NCBI BLAST+ program. BLASTN databases were constructed for all the LIRs identified in each of the 427 eukaryotic genomes. Users can choose to BLAST against any one or more BLASTN databases. The detailed steps to perform BLAST in LIRBase in shown in Figure 10.
Figure 10. Steps to BLAST in LIRBase.
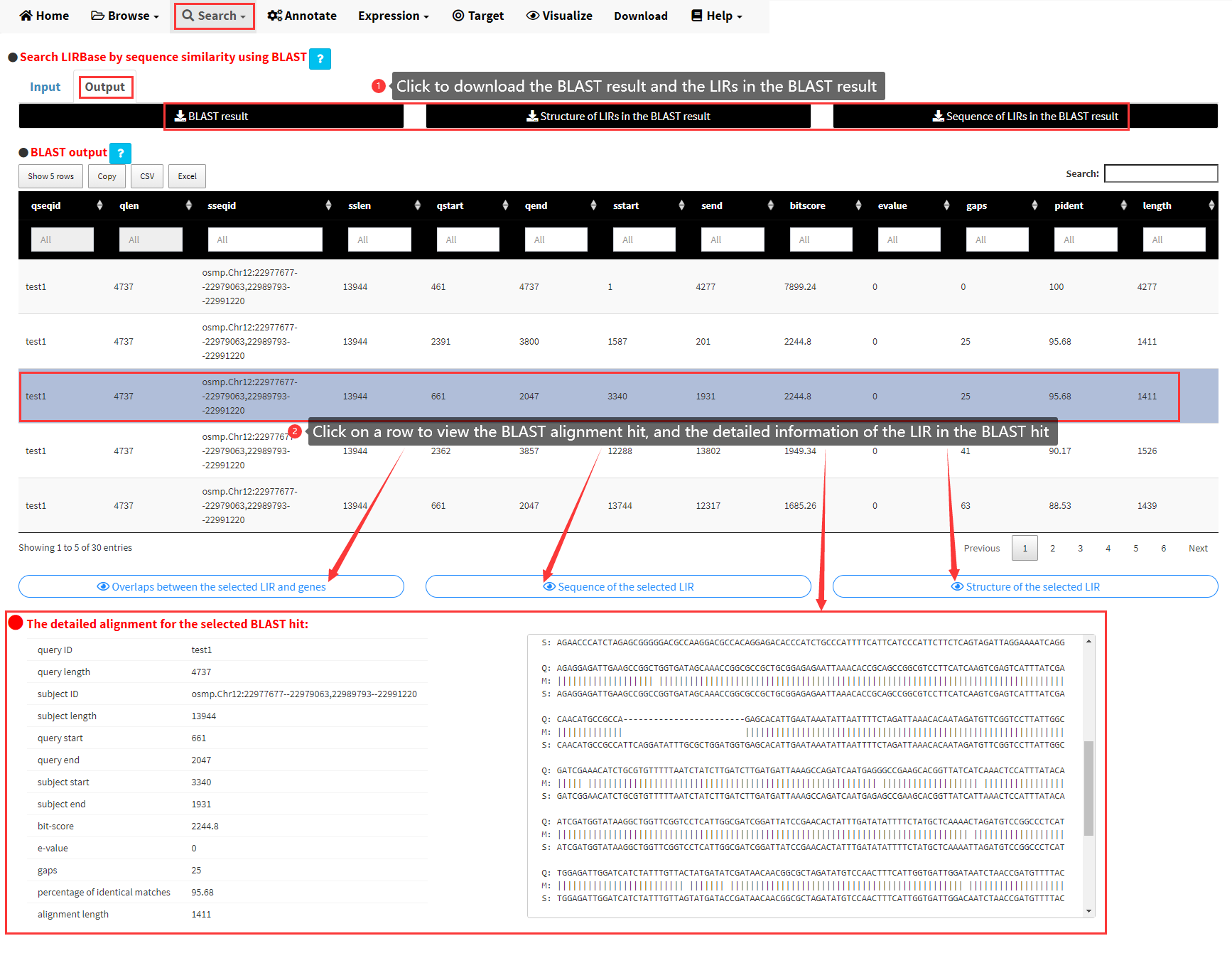
Once the BLAST alignment is finished, you would be taken to the Output panel of the Blast menu, which displays the BLAST result in a data table (Figure 11). The whole BLAST results can be downloaded by clicking the download buttons on top of the data table. Each row of the data table represents a BLAST hit. By clicking a row of this table, the detailed information of the selected BLAST hit would be displayed, including the alignment of a query sequence and a subject LIR sequence in the BLAST database. Three buttons would also be displayed below the table if any row of the table was clicked. The structure, sequence of the LIR in this BLAST hit and the overlaps between this LIR and genes in the corresponding genome, can be viewed by clicking the three buttons (Figure 11).
Figure 11. The Output panel of the Blast submenu.
5. Annotate long inverted repeats in user-uploaded DNA sequences
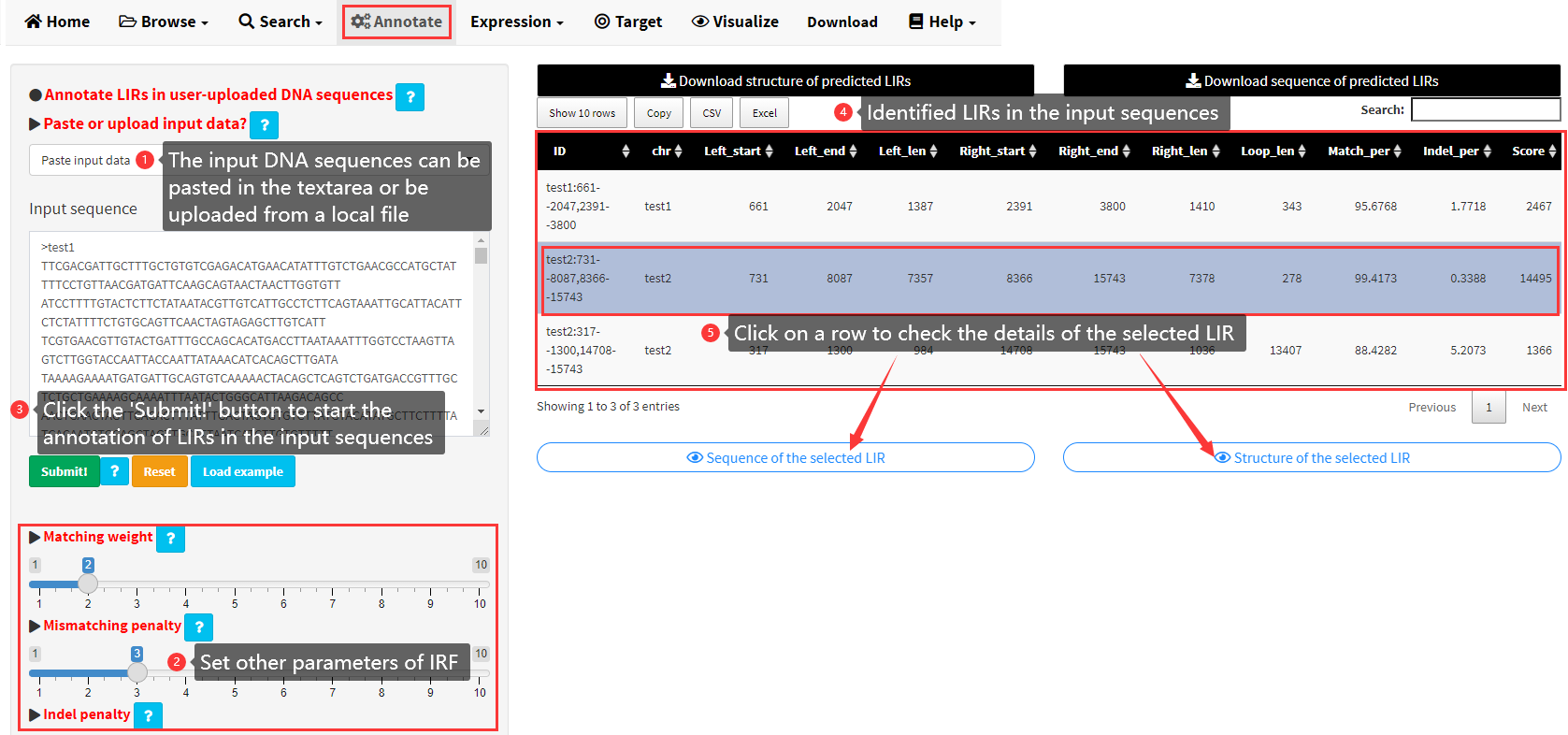
The software IRF (https://tandem.bu.edu/irf/irf.download.html) was utilized to identify long inverted repeats in the 427 eukaryotic genomes collected in LIRBase. IRF can only be used in the command line. We implemented a graphical interface for users to annotate long inverted repeats in user-uploaded DNA sequences by IRF (Figure 12). The detailed steps to annotate LIRs in user-uploaded DNA sequences are shown in Figure 12. The input DNA sequences for IRF can be pasted in a text area or be uploaded from a local text file. The input data must be DNA sequences in fasta format. Each sequence should have a unique ID start with “>”.
Figure 12. The Annotate menu of LIRBase to annotate LIRs in user-uploaded DNA sequences.
The sequences and structures of LIRs identified by IRF can be downloaded as text files (Figure 12). The result of IRF are listed in a data table (Figure 12). Each row shows the structure of an identified long inverted repeat. Two buttons would be displayed below the table if any row of the table was clicked. The detailed information of the selected LIR including the sequence, structure of the selected LIR, can be viewed by clicking the two buttons (Figure 12).
6. Identify candidate LIRs encoding long hpRNAs by aligning sRNA sequencing data to LIRs
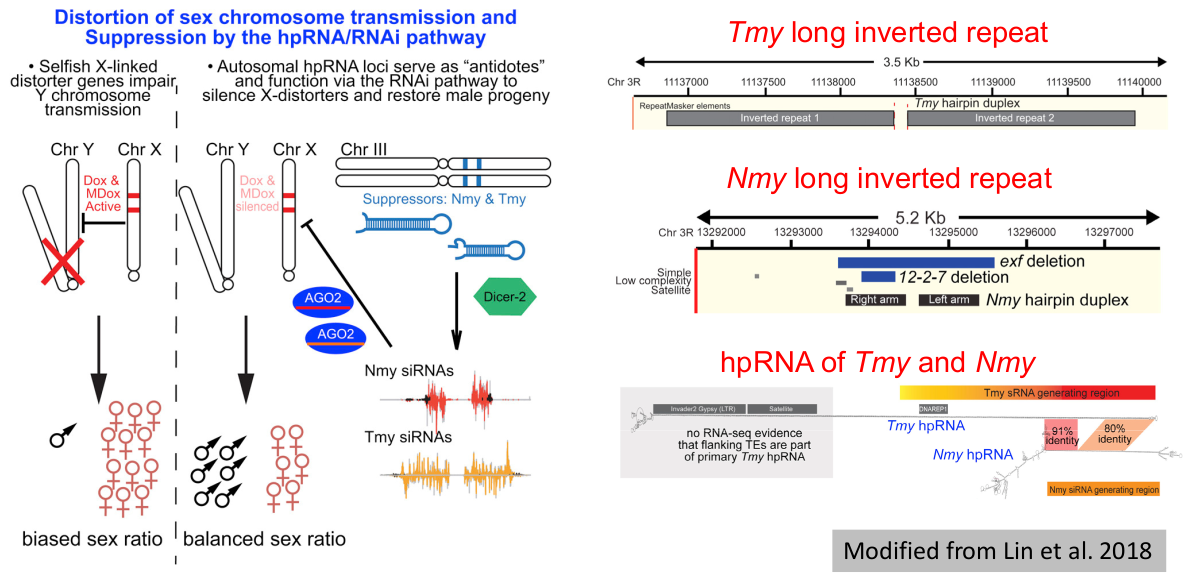
When transcribed, long inverted repeat can form long hairpin RNA genes (hpRNAs), which are much longer than typical animal or plant pre-miRNAs. Henderson et al. (2006) reported the biogenesis of small interfering RNAs (siRNAs) from long inverted repeat in Arabidopsis thaliana for the first time. This siRNA biogenesis pathway was soon reported and verified in other animals and plants.
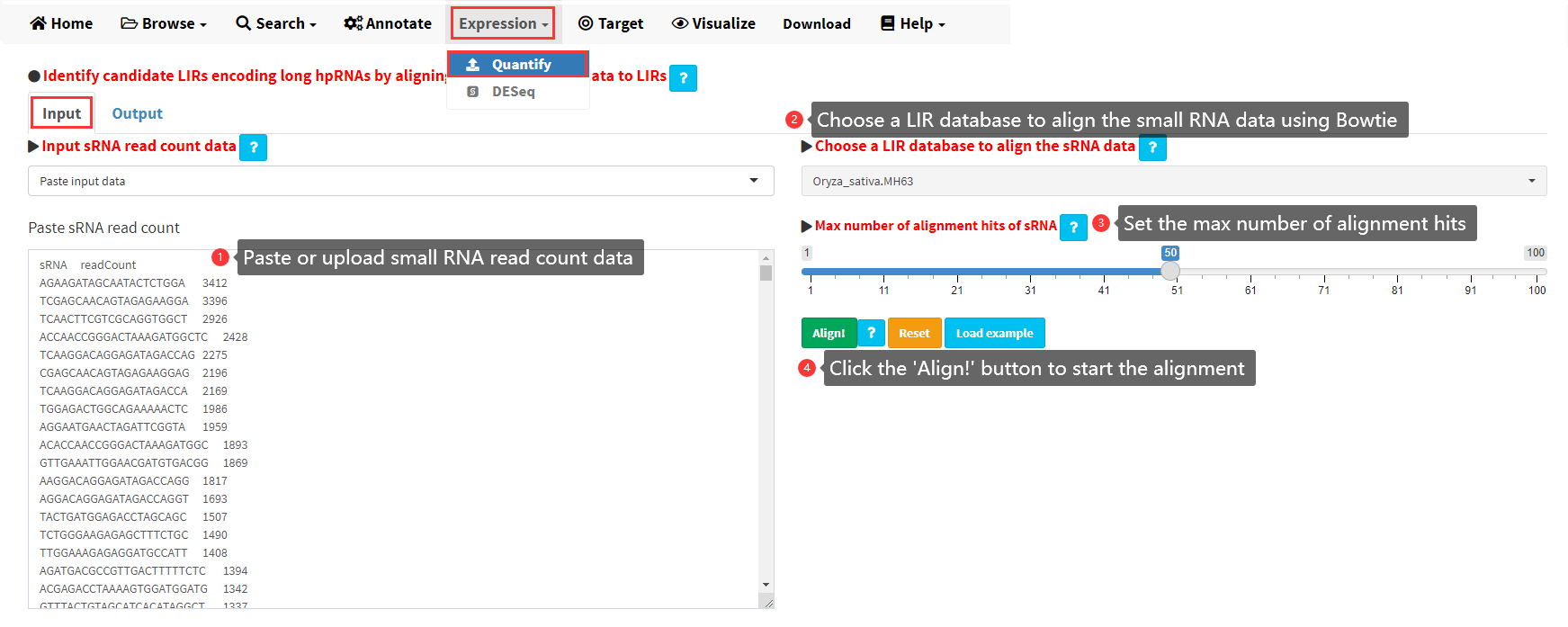
To facilitate the annotation of small RNAs derived from LIRs, we implemented a functionality in LIRBase allowing alignment of user-uploaded small RNA sequencing data to all the identified LIRs of a genome by Bowtie (Figure 13). The input data should be read count of small RNAs rather than the raw small sequencing data as shown in Figure 13. The input small RNA read count data can be pasted in a text area provided or be uploaded from a local text file.
Figure 13. The Quantify submenu under the Expression menu of LIRBase to align small RNA sequencing data to a LIR database.
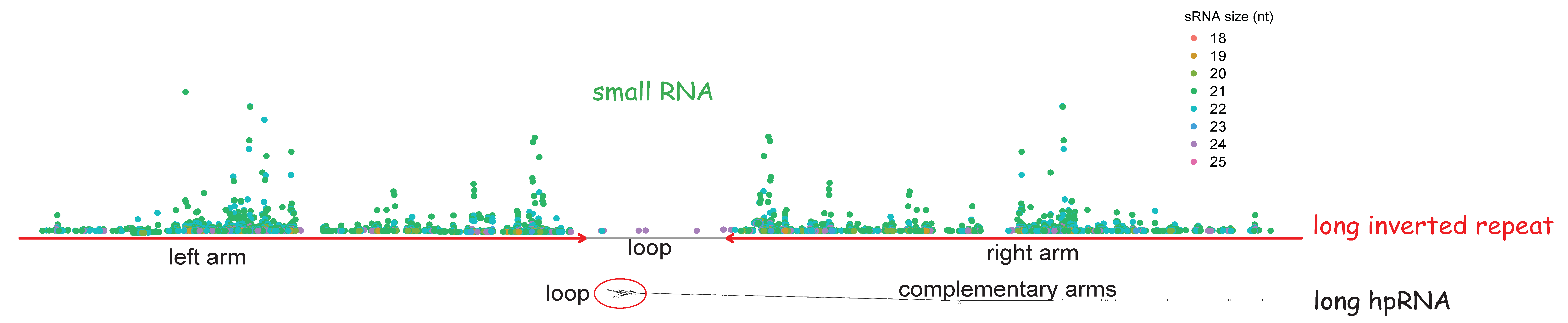
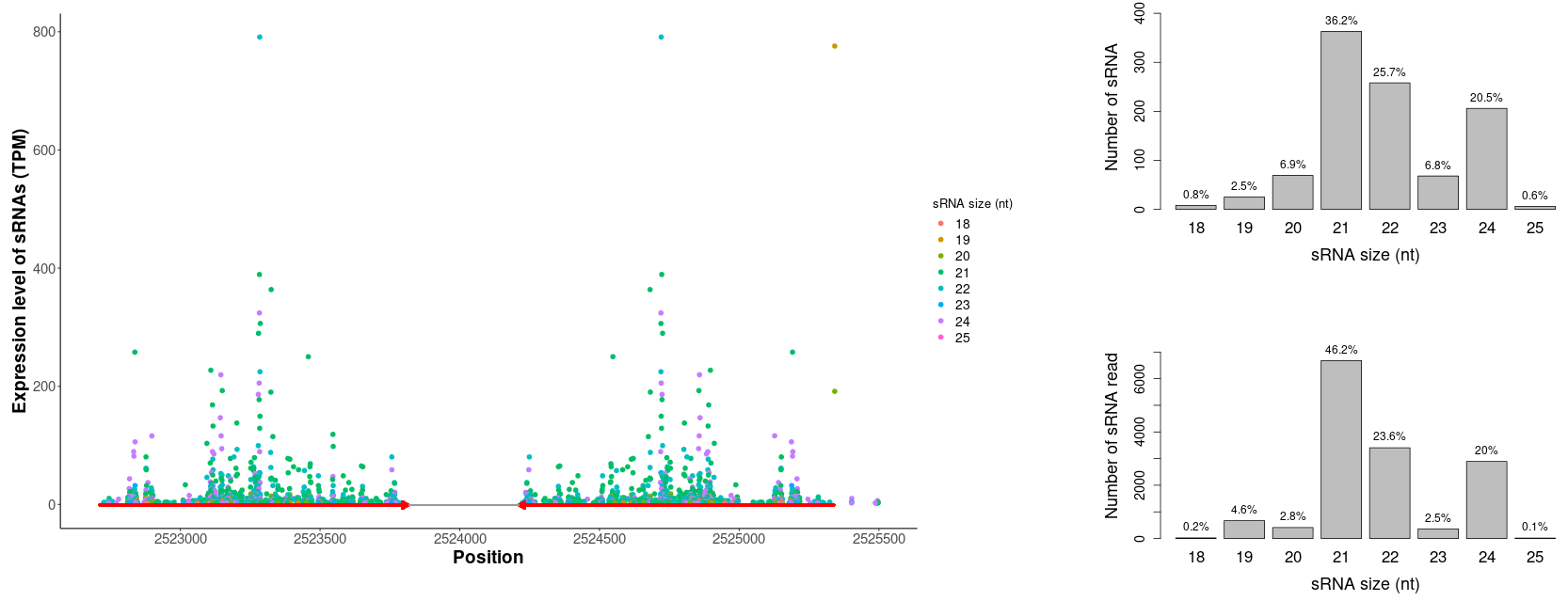
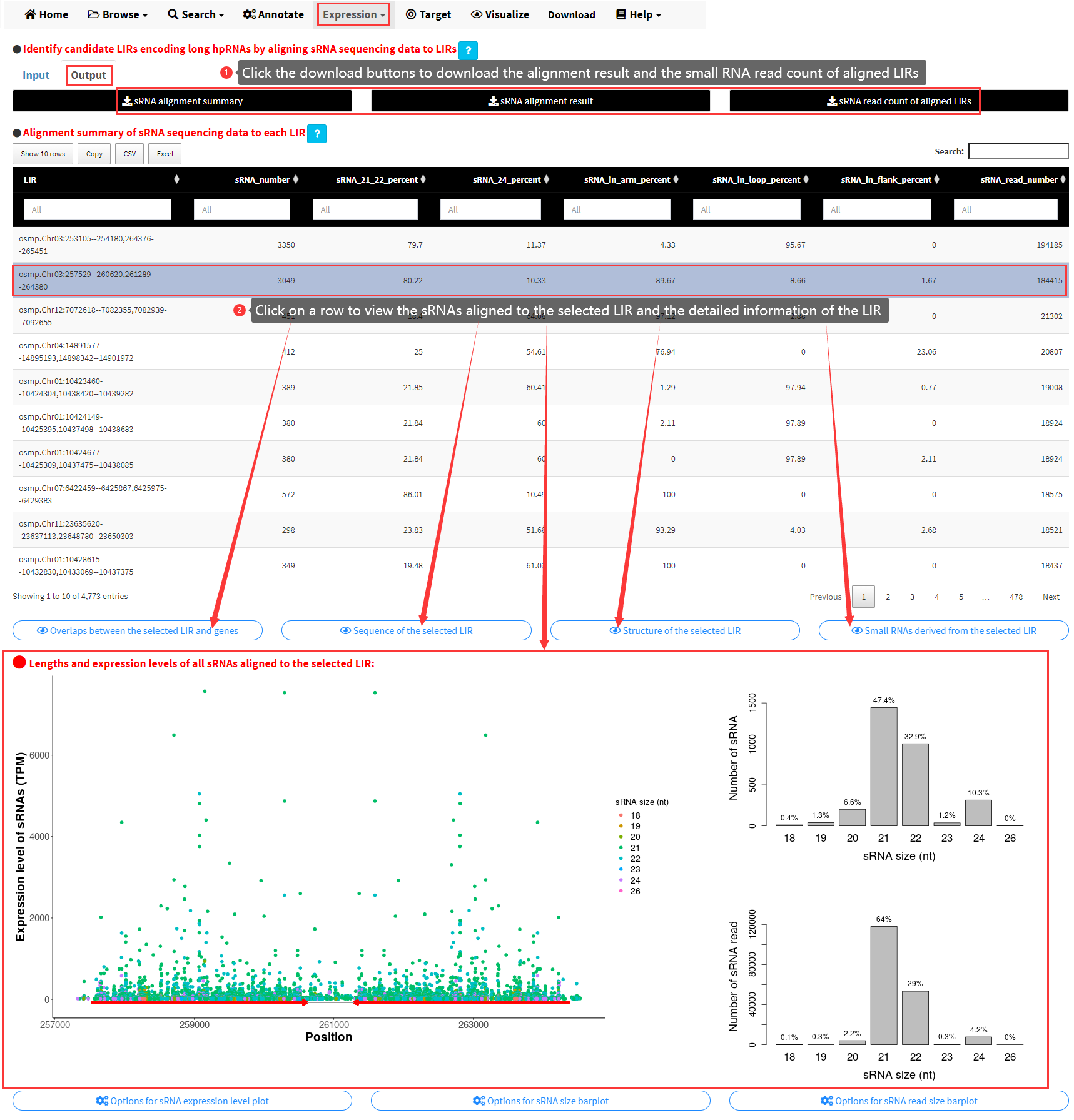
After clicking the Align! button, the alignment would be performed by Bowtie. The alignment results would be displayed in the Output panel of the Quantify submenu (Figure 14). The detailed alignment result, the summary of the alignment and the sRNA read count of aligned LIRs can be downloaded. What's more, the summary of alignment result for all aligned LIRs can be viewed as a data table. By clicking on a single row of the data table, the size distributions of sRNAs and the alignment of sRNAs to the LIR would be plotted. The detailed information of the chosen LIR would be displayed by clicking the four buttons below the table of sRNA alignment summary.
Figure 14. The Output panel of the Quantify submenu of LIRBase.
For each LIR in the data table of sRNA alignment summary, the following information are displayed as different columns.
- The number of sRNAs aligned to the LIR.
- The number of sRNA sequencing reads aligned to the LIR.
- The percentage of 21-nt and 22-nt sRNAs among all sRNAs aligned to the LIR.
- The percentage of 24-nt sRNAs among all sRNAs aligned to the LIR.
- The percentage of sRNAs aligned to the arms of the LIR among all sRNAs aligned to the LIR.
- The percentage of sRNAs aligned to the loop of the LIR among all sRNAs aligned to the LIR.
- The percentage of sRNAs aligned to the flanking sequences of the LIR among all sRNAs aligned to the LIR.
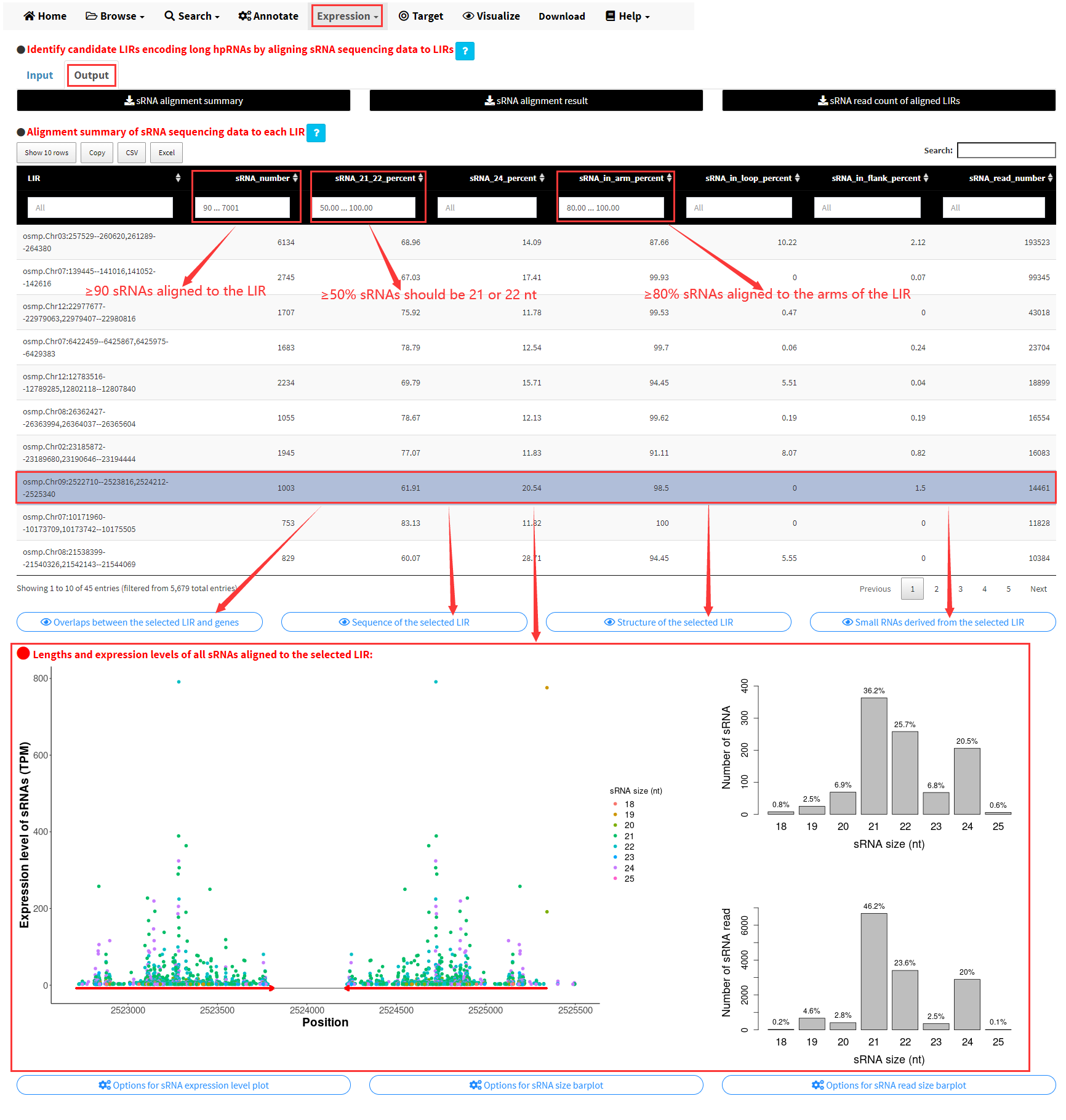
At the top of this table, we can set the values of different columns to identify candidate LIRs encoding long hpRNAs. For example, we can identify LIRs encoding candidate long hpRNAs in the genome of Minghui 63 with the following request: (1) a minimum of 90 sRNAs aligned to the LIR, (2) a minimum of 80% sRNAs aligned to the arms of the LIR, (3) a minimum of 50% sRNAs should be 21 or 22 nt (Figure 15).
Figure 15. Set the values of different columns of the table of sRNA alignment summary to identify LIRs encoding candidate long hpRNAs.
7. Differential expression analysis of long inverted repeats and small RNAs
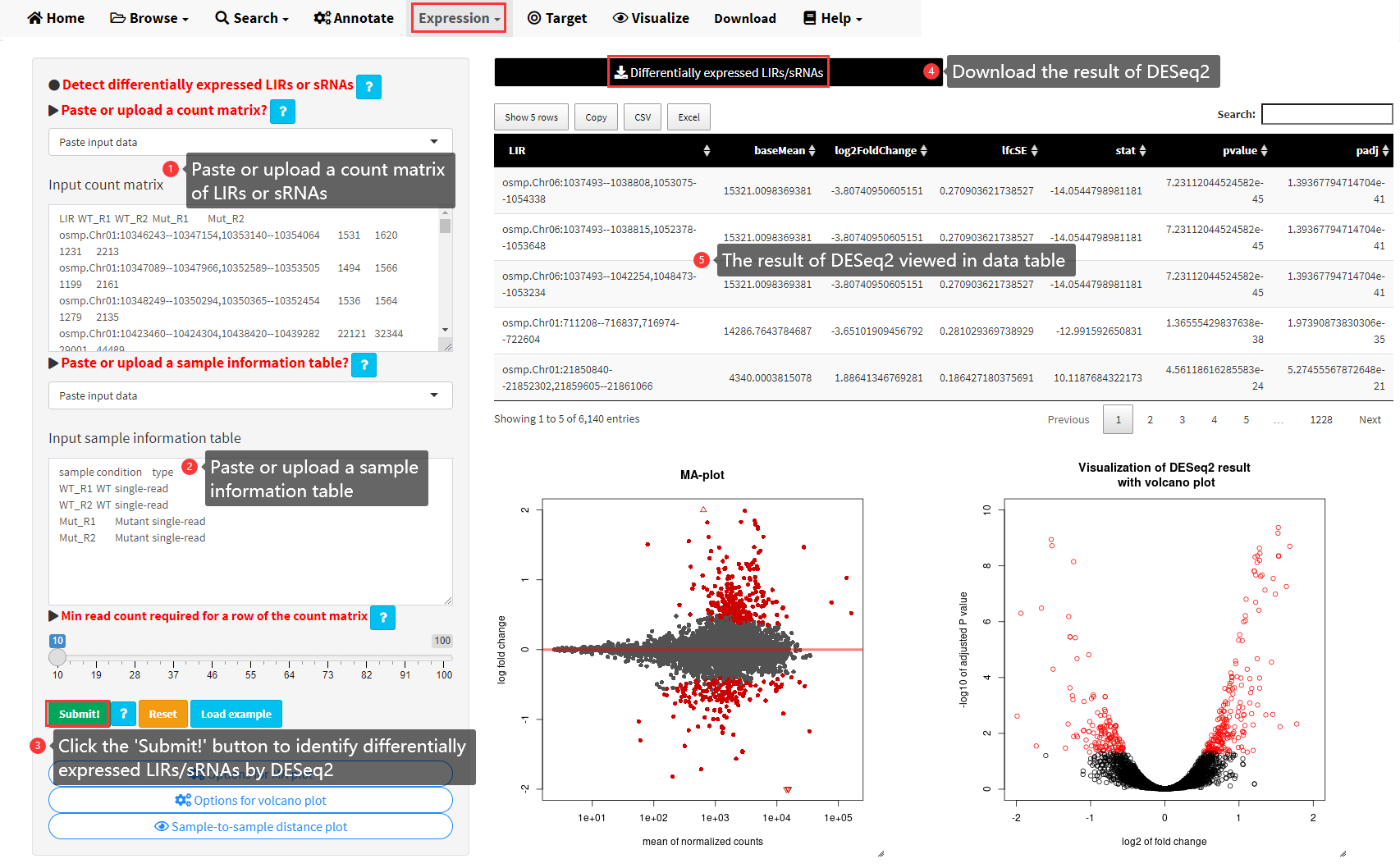
By aligning small RNA sequencing data to LIRBase, we can obtain the small RNA read count for each LIR in a genome. With multiple biological samples/tissues, we can perform differential expression analysis of long inverted repeats between different biological samples/tissues (Figure 16). The R package DESeq2 (http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html) was utilized to perform differential expression analysis. A read count matrix and a sample information table are required as input data for the differential expression analysis. The sample names in the count matrix and the sample names in the information table must be in the same order. Check the example data provided by LIRBase for the format of a sample information table.
Figure 16. The DESeq submenu under the Expression menu of LIRBase to perform differential expression analysis of LIRs/sRNAs.
The results of DESeq2 can be downloaded as a plain text file or can be viewed in a data table (Figure 16). In addition, the MA-plot and the volcano plot showing the identified differentially expressed LIRs/sRNAs are also generated. A heatmap displaying the sample-to-sample distances can be viewed by clicking the button at the bottom of the sidebar panel under the DESeq submenu.
8. Predict mRNA targets of small RNAs encoded by a LIR
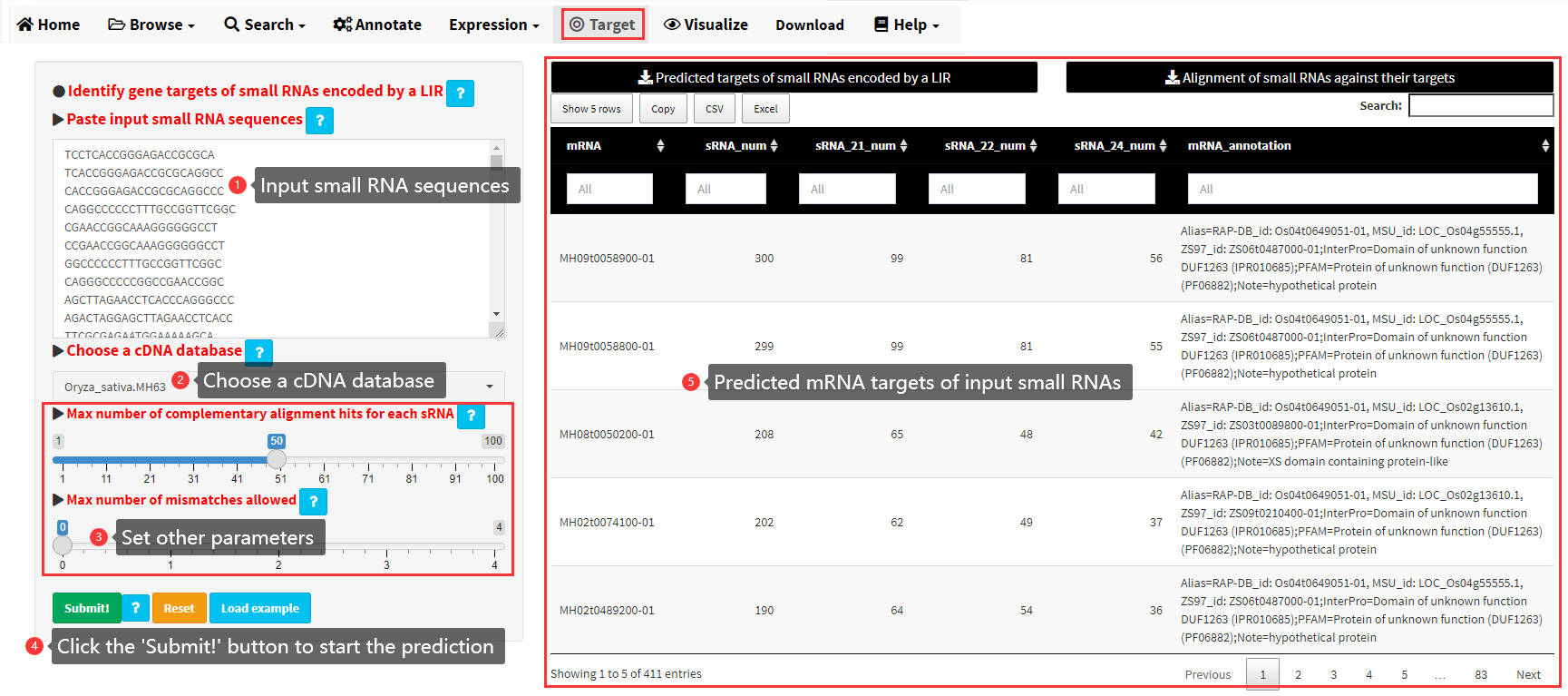
An analysis module was implemented to predict the mRNA targets of small RNAs encoded by a LIR through the detection of complementary matches between small RNAs and the cDNA sequence of protein-coding genes. The input data should be all the small RNAs encoded by a LIR in FASTA format or sequences only (Figure 17). Then the small RNA sequences can be aligned to the cDNA sequences of a specific genome by Bowtie. The alignments were processed to identify complementary matches between small RNAs and the cDNA sequences. An example output is shown in Figure 17.
Figure 17. The Target menu of LIRBase.
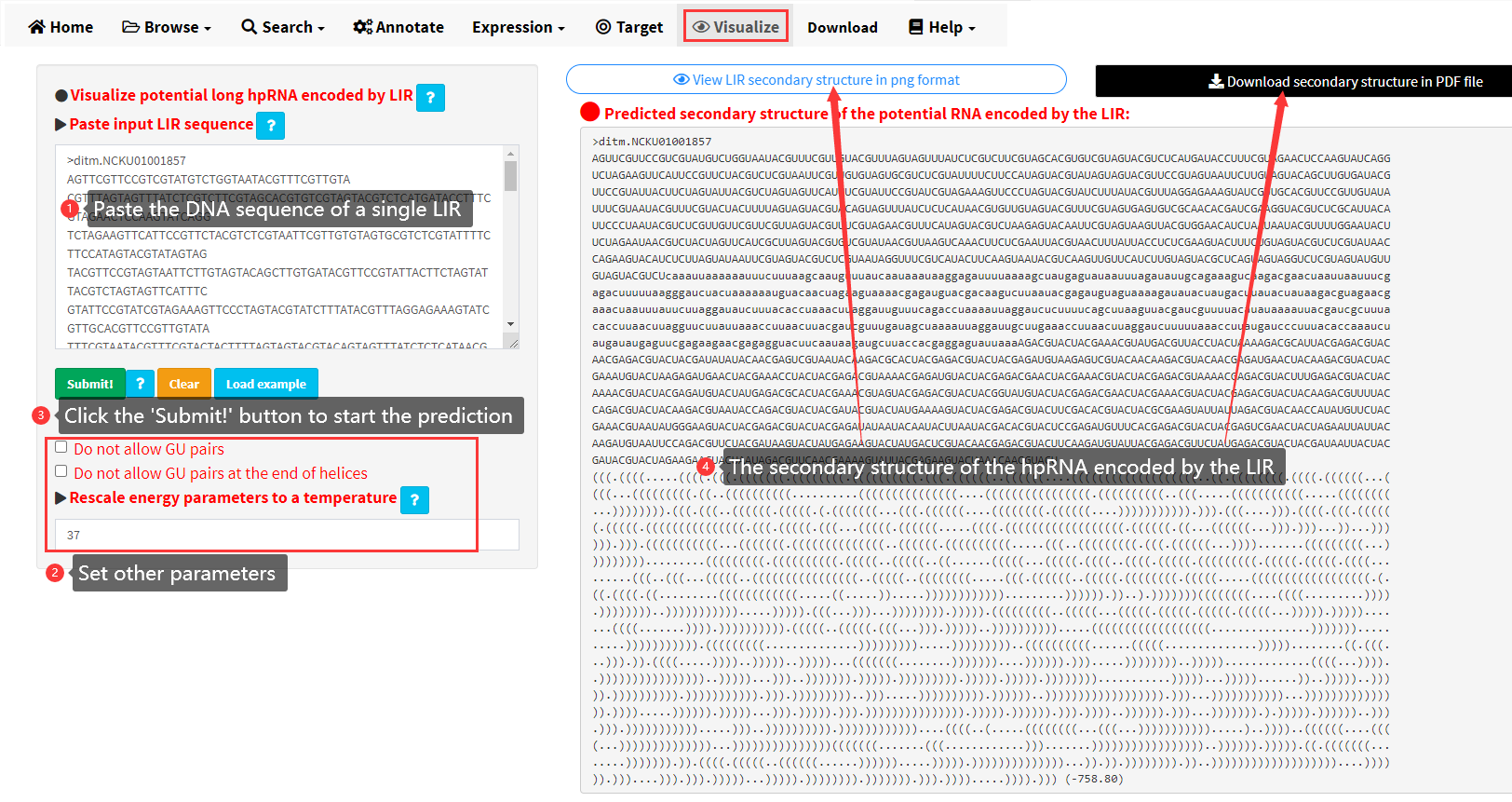
9. Predict and visualize the secondary structure of the potential hpRNA encoded by a LIR
We utilized the RNAfold software to predict and visualize the secondary structure of the potential hpRNA encoded by a LIR (Figure 18). The input data should be the DNA sequence of a single LIR. The secondary structure in dot-bracket notation is displayed in the main panel. The secondary structure in PNG image can be viewed by clicking the button on the top of the main panel. High-resolution image of the predicted secondary structure can be downloaded as PDF files.
Figure 18. The Visualize menu of LIRBase.
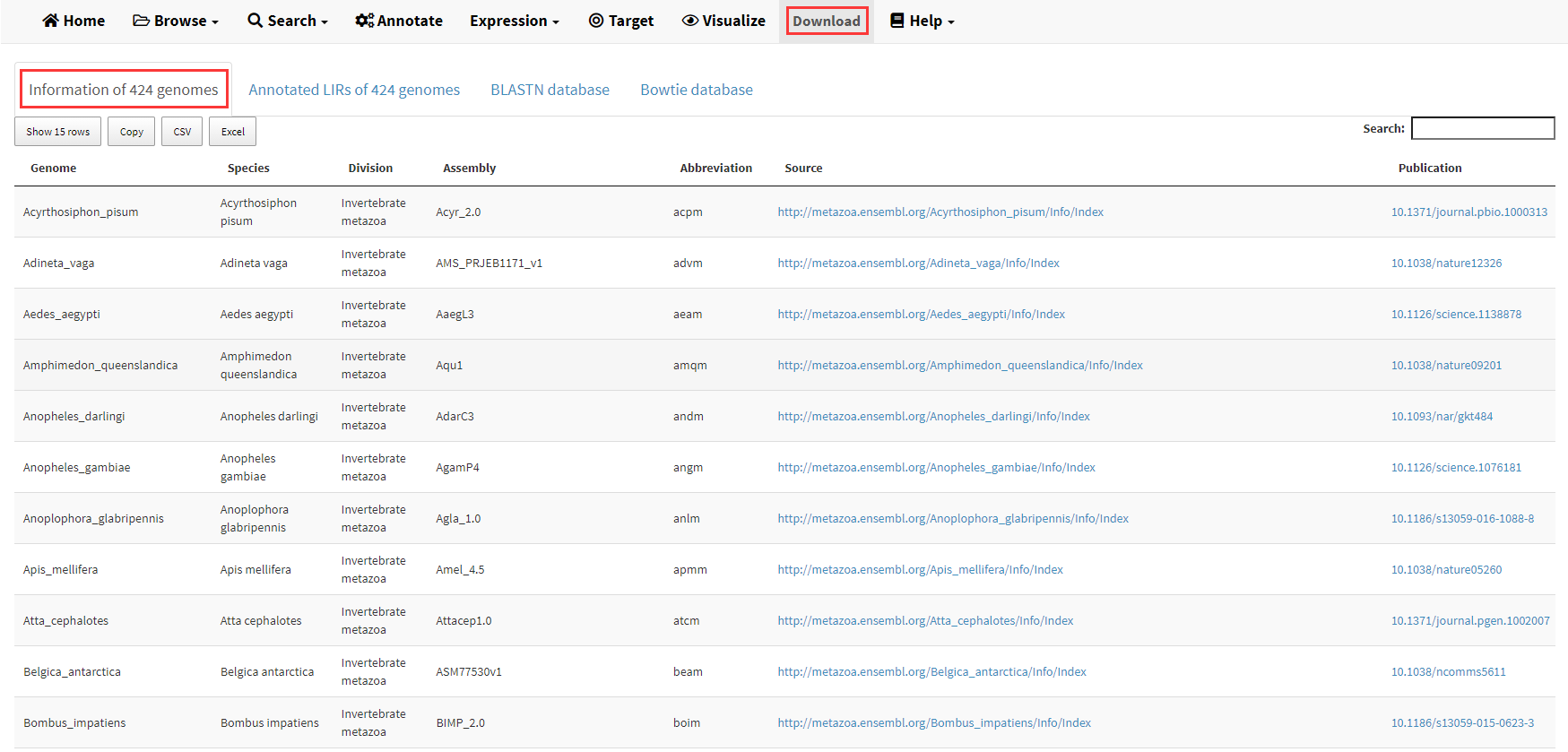
The information of 427 genomes collected in LIRBase is displayed in the Download menu of LIRBase (Figure 19).
Figure 19. The Download menu of LIRBase.
11. Download LIRs identified in 427 eukaryotic genomes, and the corresponding BLAST/Bowtie index database
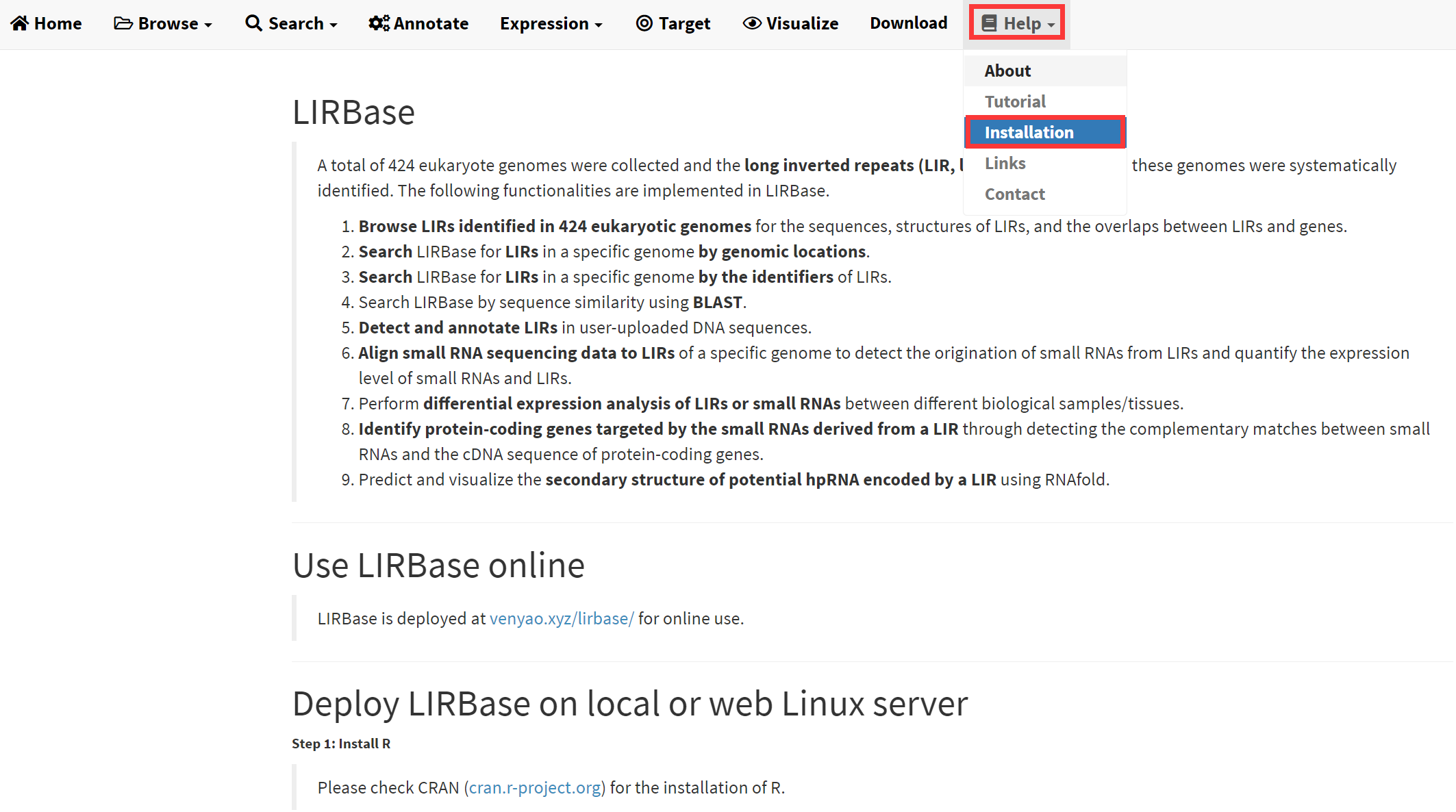
In addition to be used online at https://venyao.xyz/lirbase/, LIRBase can be deployed on a personal local or web Linux server. Deployment of LIRBase is platform independent, i.e., LIRBase can be deployed on any platform with the R environment available. The detailed steps are described in the Installation submenu under the Help menu of LIRBase (Figure 20).
Figure 20. The Installation submenu under the Help menu of LIRBase.
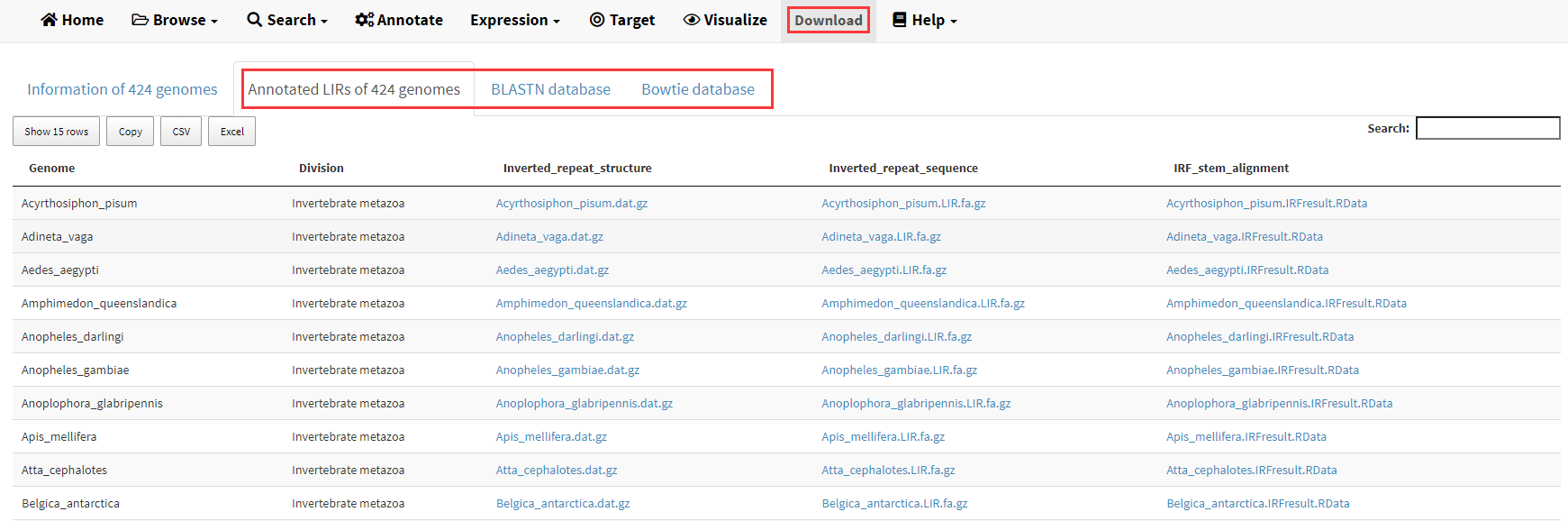
The source code of LIRBase is deposited in GitHub (https://github.com/venyao/LIRBase). As the file size of identified LIRs and the corresponding BLAST/Bowtie databases of the 427 eukaryotic genomes are too large, these datasets were not deposited in GitHub. Instead, these data can be downloaded from https://venyao.xyz/lirbase/ through the Download menu (Figure 21).
Figure 21. The Download menu of LIRBase.
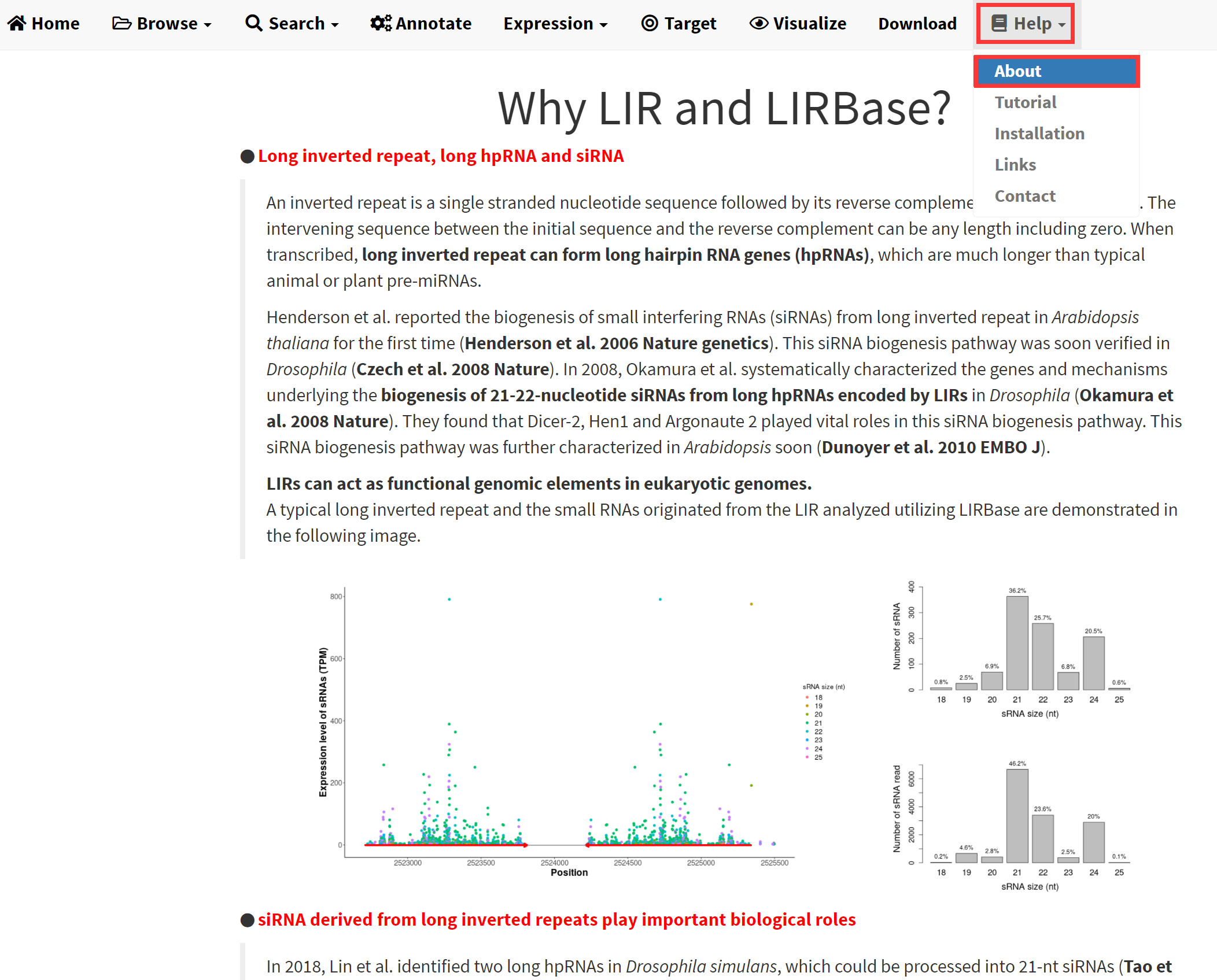
12. About LIR and LIRBase
The definition of long inverted repeat, the biogenesis pathway of siRNAs from long inverted repeat and the biological roles of siRNAs generated in this pathway are elaborated in the About submenu under the Help menu of LIRBase (Figure 22). These results implied that a platform for comprehensive annotation and analysis of siRNAs derived from long inverted repeat is in urgent need.
Figure 22. The About submenu under the Help menu of LIRBase.